




Содержание
Перейти к:
https://doi.org/10.24060/2076-3093-2025-15-2-28-42
Перейти к:
Агрессивная и устойчивая к терапии природа глиобластомы делает ее одним из самых смертельных злокачественных новообразований у людей. Полная хирургическая резекция сложна, и для лечения оставшихся опухолевых клеток за пределами границы опухоли используется комбинация химио- и лучевой терапии, вызывающая повреждение ДНК опухолевой клетки и активирующая пути апоптоза. К сожалению, рецидивы являются обычным явлением и серьезным препятствием в лечении, часто встречающимся при более агрессивной и устойчивой к лечению глиобластоме. Известно, что репарация или восстановление ДНК и сигнальные пути повреждения ДНК имеют решающее значение для поддержания геномной стабильности. Дефекты путей репарации ДНК и сигнализации повреждения способствуют возникновению опухолей, но также делают опухолевые клетки уязвимыми к повреждению ДНК и зависимыми от остаточной репарационной и сигнальной активности. Понимание молекулярных элементов этих механизмов и выявление потенциальных терапевтических/фармакологических мишеней стали важнейшими задачами для эффективного лечения пациентов с глиобластомой. Доказано, что субпопуляция стволоподобных клеток, обозначенная как опухолевые стволовые клетки (ОСК) глиобластомы, ответственны не только за возникновение, поддержание и рецидив опухоли, они же поддерживают устойчивость к химиолучевой терапии из-за их повышенной способности к восстановлению ДНК. Более того, есть доказательства связей между углеводным метаболизмом и путями восстановления ДНК, что может открыть новые терапевтические возможности при глиобластоме. В данной работе обсуждаются современные стратегии изучения молекулярных механизмов целенаправленных путей репарации повреждения ДНК при глиобластоме. Мы суммируем недавний прогресс в наших знаниях о путях и факторах, вовлеченных в устранение повреждений ДНК, вызванных ионизирующим излучением и темозоломидом (TMZ) в частности. Наконец, мы представляем терапевтические стратегии, основанные на ингибиторах путей репарации ДНК, которые в настоящее время тестируются в преклинических или в клинических испытаниях.
Гареев И.Ф., Бейлерли О.А., Румянцев С.А. Повреждение и восстановление ДНК при глиобластоме: новые перспективы терапии. Креативная хирургия и онкология. 2025;15(2):124-138. https://doi.org/10.24060/2076-3093-2025-15-2-28-42
Gareev I.F., Beylerli O.A., Roumiantsev S.A. DNA Damage and Repair in Glioblastoma: Emerging Therapeutic Perspectives. Creative surgery and oncology. 2025;15(2):124-138. (In Russ.) https://doi.org/10.24060/2076-3093-2025-15-2-28-42
Глиобластома (глиома 4-й степени по классификации Всемирной организации здравоохранения (ВОЗ) 2021 года) представляет собой наиболее частую и агрессивную злокачественную первичную опухоль головного мозга у взрослых. В плане терапии пациенты с глиобластомой обычно подвергаются максимальной резекции опухолевой массы с последующей одновременной лучевой и химиотерапией с использованием алкилирующего агента темозоломида (TMZ) [1]. Эти методы лечения, хотя и являются стандартными, однако сталкиваются со значительными трудностями, особенно из-за способности опухоли широко проникать в окружающие ткани мозга и наличия гематоэнцефалического барьера (ГЭБ), который ограничивает эффективность многих системных методов лечения [2]. Кроме того, гетерогенность глиобластомы и высокая частота рецидивов создают дополнительные препятствия для лечения [2]. Поэтому, изучение и получение новой информации о молекулярных механизмах развития и прогрессирования глиобластомы, а также о задействованных в них сигнальных путях могут иметь решающее значение для разработки новых терапевтических методов. Устойчивость к лучевой и химиотерапии характерна для многих типов злокачественных новообразований; однако неясно, приобретается ли устойчивость во время прогрессирования опухоли или она заранее связана с генетическими изменениями, которые изначально приводят к развитию опухоли.
Повреждение ДНК от воздействия различных факторов окружающей среды, а также эндогенных токсичных агентов, таких как свободные радикалы, может поставить под угрозу стабильность генома и вызвать или способствовать возникновению многих заболеваний, в том числе опухолей. Поскольку ДНК является основным генетическим материалом, она жизненно важна для обеспечения целостности структуры и функции клеток для поддержания нормальной жизнедеятельности и стабильных характеристик вида. Действительно, при воздействии эндогенных или экзогенных стрессов клетки могут генерировать различные типы повреждений ДНК. Распространенные экзогенные факторы, такие как токсичные тяжелые металлы и ионизирующее излучение, были хорошо изучены и, как было установлено, вызывают серьезные повреждения ДНК. Эндогенные вещества часто высвобождаются во время метаболизма экзогенных веществ в организме или после повреждения клеток и потери целостности клеточной мембраны. Повреждение ДНК может происходить двумя путями, а именно прямыми и косвенными эффектами. В прямом пути эндогенные или экзогенные вещества напрямую контактируют с ДНК, что приводит к разрыву химических связей в молекулах ДНК и, таким образом, изменению структуры и активности ДНК [3][4].
Пути репарации или восстановления ДНК являются одними из важнейших ключевых игроков онкогенных мутаций в глиобластоме, связанных с устойчивостью как к химио-, так и к лучевой терапии. Например, наиболее распространенными изменениями путей репарации ДНК в глиобластоме являются снижение регуляции сигнальных путей p53, снижение регуляции сигнальных путей ретинобластомы и метилирование промотора O6-метилгуанин-ДНК-метилтрансферазы (MGMT) (рис. 1) [5–7].
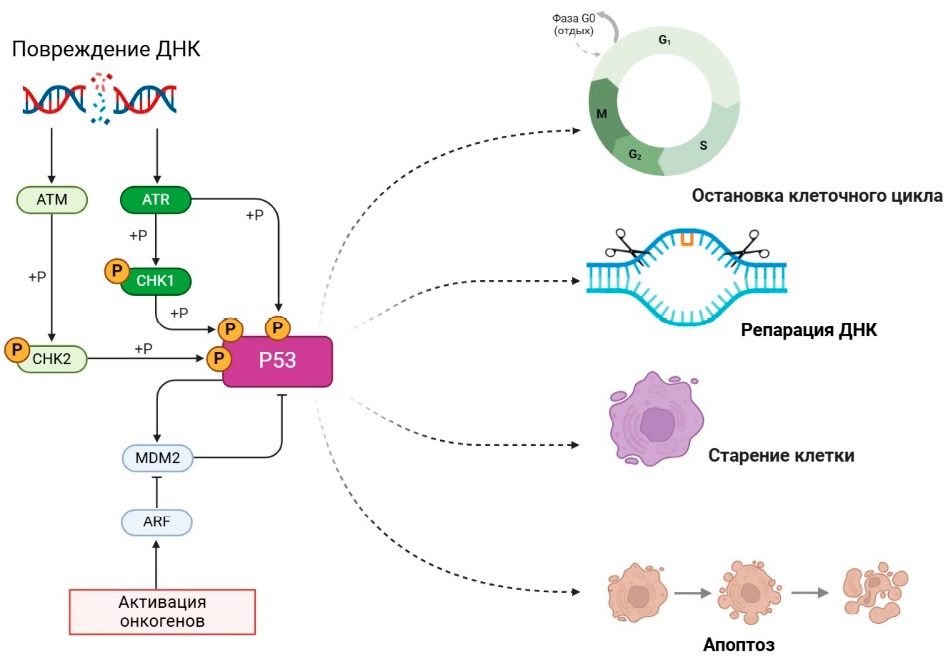
Рисунок 1. p53 Регулирование и сигнализация
Регуляция и сигнализация p53 включают контроль и передачу сигналов, опосредованных белком — супрессором опухолей p53. p53 регулирует прогрессирование клеточного цикла, восстановление ДНК и апоптоз для поддержания геномной стабильности и предотвращения образования опухолей. Нарушение регуляции функции P53 часто встречается при опухолях (в том числе глиобластоме), что приводит к неконтролируемому росту клеток. Исследование механизмов регуляции и сигнализации p53 дает ценные знания для разработки целевых терапевтических подходов против опухоли
Figure 1. p53 regulation and signaling
The tumor protein p53 regulates cell cycle progression, DNA repair, and apoptosis, thereby maintaining genome stability and preventing tumor formation. The dysregulation of p53 function is common in tumors, including glioblastoma, leading to uncontrolled cell proliferation. Research into p53 regulation and signaling provides critical insights for the development of targeted anti-tumor therapies
Кроме того, инактивация сигнальных путей фосфатазы с двойной субстратной специфичностью (PTEN) и активация рецепторов эпидермального фактора роста (EGFR)/фосфоинозитид-3-киназы (PI3K) сигнального пути, которые обнаружены примерно в одной трети случаев у пациентов с глиобластомой, как считается, усиливают пути ответа на повреждение ДНК в глиобластоме [8]. Высокая частота этих изменений в глиобластоме предполагает, что пути репарации повреждений ДНК играют важную роль в онкогенезе глиобластомы и что поиск новых сигнальных путей, ответственных за глиомагенез, может обеспечить новые подходы к эффективной терапии для пациентов с глиобластомой.
Химио- и лучевая терапия напрямую или косвенно вызывают гибель клеток через повреждение ДНК, и на успех терапии влияют несколько биохимических путей. Кроме того, генетический фон значительно влияет на результат лечения. Клеточный ответ включает сложный сигнальный каскад, называемый реакцией на повреждение ДНК (DDR), который отвечает за распознавание, сигнализацию и исправление повреждения ДНК. Различные виды повреждений, образующихся в ДНК, требуют специфических путей репарации повреждений ДНК, которые позволяют устранить повреждения и могут способствовать радио- и химиорезистентности глиобластомы [9]. В этой связи было разработано множество подходов к лечению, направленных на новые молекулярные мишени, которые можно использовать в качестве терапевтических альтернатив. Тем не менее большинство из них терпят неудачу во время клинических испытаний, что свидетельствует о том, что единая стратегия нацеливания не улучшает терапевтические результаты. Неудачи, связанные с этими подходами, могут быть связаны с компенсаторными механизмами DDR, высокой системной токсичностью, отсутствием стабильности препаратов и недостаточностью исследований in vitro и in vivo, демонстрирующих эффективность новых препаратов [10][11].
Различные эндогенные и экзогенные агенты, повреждающие ДНК, такие как ионизирующее излучение и химиотерапевтические агенты, могут приводить к повреждениям ДНК, включая одноцепочечные разрывы (SSB) и двухцепочечные разрывы (DSB), химические модификации оснований или сахаров, а также межцепочечные или внутрицепочечные сшивки [12]. Если повреждение ДНК не исправить, оно вызовет геномную нестабильность и мутацию, что является одним из признаков онкогенеза. Чтобы предотвратить эту ситуацию, в процессе эволюции клетки млекопитающих выработали ряд механизмов, называемых DDR, для борьбы с такими повреждениями. DDR — это сложная сеть, которая функционирует по-разному, чтобы воздействовать на различные повреждения ДНК, включая передачу сигнала, регуляцию транскрипции, контрольные точки клеточного цикла, индукцию апоптоза, процессы толерантности к повреждениям и множественные пути восстановления ДНК. Пути восстановления повреждений ДНК имеют два противоположных аспекта: с одной стороны, они защищают целостность генетического материала нормальных клеток, с другой стороны, они способствуют устойчивости опухолевых клеток к генотоксической терапии [3][4]. В начале формирования опухоли механизм DDR постоянно активируется репликацией, вызванной онкогенами, и окислительным стрессом и действует как защитный механизм, предотвращающий распространение злокачественных клонов; однако во время трансформации опухолевые клетки могут накапливать и переносить повреждения генома и перестройки из-за аберраций DDR [13]. Поскольку системы репарации ДНК снижают эффективность генотоксических методов лечения, понимание и характеристика механизмов репарации имеют первостепенное значение для разработки новых терапевтических стратегий [13].
Известно, что в клетках млекопитающих двумя основными органеллами, содержащими ДНК, являются ядро и митохондрии. Системы восстановления ядерной ДНК делятся на следующие основные пути: 1) прямая реверсия, которая в основном восстанавливает повреждения, вызванные алкилирующими агентами; 2) эксцизионная репарация оснований (BER), направленная на SSB и необъемные поврежденные основания ДНК; 3) эксцизионная репарация нуклеотидов (NER), исправляющая объемные, искажающие спираль повреждения ДНК; 4) репарация ошибочно спаренных нуклеотидов (MMR); 5) рекомбинационная репарация, которая далее подразделяется на репарацию посредством гомологичной рекомбинации (HRR) и негомологичное соединение концов (NHEJ), в основном функционирующие при DSB; 6) альтернативное негомологичное соединение концов (alt-NHEJ), участвующее в восстановлении DSB; 7) транслезионный синтез, который, скорее всего, является механизмом устойчивости к повреждению ДНК. Пути восстановления митохондриальной ДНК, включая прямую репарацию, BER, MMR, транслезионный синтез и репарацию DSB, могут восстанавливать поврежденную ДНК, поддерживая генетическую целостность митохондрий, защищая митохондриальную ДНК от окислительного повреждения и способствуя выживанию клеток (рис. 2) [14–16].
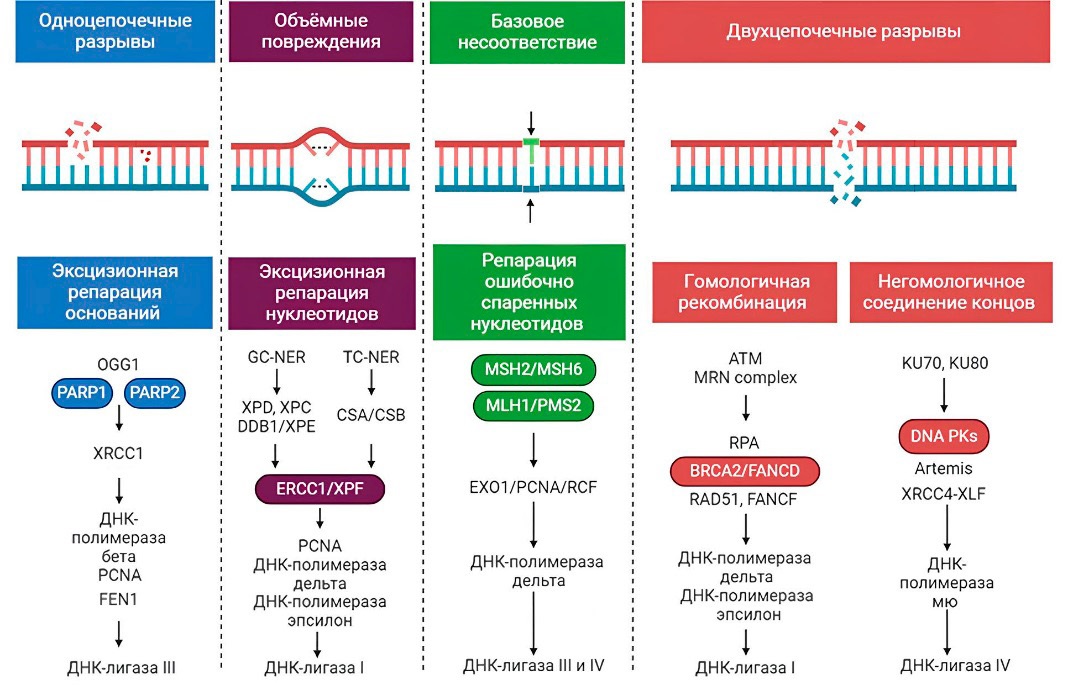
Рисунок 2. Механизмы восстановления или репарации ДНК после ее повреждения
Figure 2. DNA repair mechanisms following DNA damage
Пути репарации ДНК играют важную роль в поддержании стабильности и целостности генома посредством исправления поврежденной ДНК, которая может способствовать запуску онкогенеза. Многочисленные исследования показали, что некоторые виды опухолей, в том числе глиобластома, связаны с дефектом или мутацией в белках ядерных или митохондриальных путей репарации ДНК [17]. Люди, которые являются носителями мутации гена MMR, имеют повышенный риск развития самых разных видов опухолей, чем их родственники, не являющиеся носителями. Например, два важных гена, связанных с репарацией ДНК HRR, ген рака молочной железы 1 (BRCA1) и ген рака молочной железы 2 (BRCA2), обусловливают генетическую предрасположенность к раку молочной железы, раку яичников и раку поджелудочной железы [18]. Кроме того, микроокружение опухоли, где характерно наличие гипоксии, низкого pH и дефицита питательных веществ, может привести к геномной нестабильности и прогрессированию опухоли посредством подавления пути репарации ДНК [19]. Сообщалось, что условия гипоксии могут привести к снижению экспрессии mutL гомолог 1 (MLH1), основного белка в пути MMR [20]. Понижение экспрессии RAD51 (ключевой медиатор HRR), вызванное гипоксией, наблюдалось во многих типах опухолевых клеток, что позволяет предположить, что гипоксическое микроокружение опухоли может подавлять путь HRR, вызывая генетическую нестабильность [20]. Недавние исследования показали, что внеклеточные питательные вещества оказывают значительное влияние на целостность генома. Глутамин является основным источником углерода и азота для опухолевых клеток. Недостаток глутамина приводит к повреждению алкилирования ДНК путем ингибирования активности альфа-кетоглутарат-зависимой диоксигеназы (ALKBH) и увеличения чувствительности опухолевых клеток к алкилирующим агентам. Глюкозное голодание также усиливает чувствительность к лучевой терапии опухолевых клеток путем снижения репарации DSB [21][22]. Таким образом, нарушение регуляции путей репарации ДНК может способствовать развитию опухоли, способствуя геномной нестабильности и мутации в клетках млекопитающих.
Известно, что непосредственная роль DDR в онкогенезе и резистентности глиобластомы к стандартной терапии тесно зависит от сроков оценки и типа повреждения ДНК. Более того, в начале своего формирования DDR может останавливать экспансию опухолевых клеток. Однако когда опухолевые клетки и опухолевая ниша глиобластомы установлены, DDR способствует выдержке геномной нестабильности и исправлению повреждений ДНК, вызванных влиянием химиопрепаратов и лучевой терапии [23]. Как лучевая, так и химиотерапия может быть нацелена на создание условий прямого повреждения ДНК, вызывающего непосредственно гибель опухолевых клеток. Сложный клеточный каскад активируется в ответ на различные повреждения ДНК, чтобы опосредовать клеточные изменения (например, остановку клеточного цикла) и напрямую восстановить повреждение ДНК. К тому же клетки организма человека активируют различные механизмы восстановления ДНК в зависимости от клеточного контекста и типа субстрата или повреждения, которые необходимо исправить. Механизмы репарации ДНК способствуют выживанию опухолевых клеток и связаны с процессом резистентности к существующей терапии и рецидивом глиобластом.
Существуют три основных пути репарации повреждений ДНК, которые обрабатывают повреждения алкилирования TMZ: прямая репарация MGMT, BER и MMR [14–16]. MGMT является основным ферментом, ответственным за устойчивость глиобластомы к TMZ. Экспрессия MGMT коррелирует с устойчивостью к TMZ, в основном потому, что MGMT удаляет метильную группу из О6-метилгуанина (O6-MG), восстанавливая целостность гуаниновых оснований в ДНК. Преимущества алкилирующих агентов в значительной степени ограничены пациентами, чьи опухоли показывают метилирование промотора MGMT [24]. Если MGMT не исправляет неправильное включение тимина, которое произошло во время репликации O6-MG, активируется путь MMR. Этот процесс входит в «бесполезный цикл», который заменяет неправильно включенный тимин другим тимином, что приводит к энергозатратным циклам, остановке репликативной вилки и разрывам ДНК [25]. Преобразование ошибок неправильного включения в DSB активирует пути восстановления DSB, и, если восстановление не удается, запускается апоптоз. Большинство повреждений, вызванных TMZ, таких как повреждения N3-метиладенина и N7-метилгуанина, в первую очередь восстанавливаются путем BER. Следовательно, функциональный путь BER способствует устойчивости к TMZ и связан с худшим прогнозом при глиобластоме. Помимо повреждений алкилирования, окислительные повреждения в основаниях ДНК обычно восстанавливаются BER [26][27].
В отличие от поражений, связанных с TMZ, лучевая терапия вызывает множественные типы повреждений ДНК, включая повреждение нуклеиновой кислоты, сахара и фосфатного остова. В конечном счете эти повреждения, если их не восстановить, преобразуются в DSB. Как упоминалось ранее, DSB являются высокотоксичными радиационно-индуцированными повреждениями ДНК, и их восстановление может вызвать геномные перестройки и мутации или апоптоз. Ионизирующее излучение оказывает как прямое, так и косвенное воздействие на ДНК. Прямое воздействие заключается в том, что ДНК повреждается путем прямого поглощения энергии излучения, тогда как косвенное воздействие заключается в том, что другие молекулы вокруг ДНК поглощают энергию излучения и производят аномально активные свободные радикалы, которые взаимодействуют с ДНК и другими крупными молекулами, вызывая повреждение [28][29].
Когда излучение проходит через генетический материал, отложение энергии вызывает обширное повреждение ДНК, и этот тип повреждения имеет форму DSB. Этот вид повреждения ДНК может представлять непреодолимый барьер для адаптации клеток глиобластомы от апоптоза. Однако для борьбы с данным типом повреждения ДНК был разработан сложный и точный набор регуляторных механизмов, в первую очередь многочисленные пути репарации, такие как репарация несоответствий, репарация удаления оснований, репарация удаления нуклеотидов и репарация DSB. NHEJ и HRR являются двумя ключевыми модальностями репарации DSB [16]. Контрольные точки повреждения ДНК активируются одновременно, что задерживает начало митоза и обеспечивает больше времени для репарации ДНК.
В ходе эволюции клеток глиобластомы множественные интегрированные молекулярные сигнальные пути приводят к повышению устойчивости опухолевых клеток к лучевой терапии. Поэтому понимание того, как клетки глиобластомы активируют и реализуют пути восстановления повреждений ДНК, имеет решающее значение для предотвращения восстановления ДНК опухолевых клеток и, таким образом, индукции некроза и апоптоза клеток глиобластомы. Датчики повреждения ДНК, такие как ATRIP, Rad24p, γH2AX, NBS1, BRCA1/2, Ku70/80 и РНК-полимераза, распознают сигналы повреждения, привлекают основную киназу ответа на DDR мутировавшую атаксию-телеангиэктазию» (ATM), связанную с ATM и Rad3 (ATR), ДНК-зависимую протеинкиназу (DNA-PK) и другие регуляторные факторы к местам разрыва ДНК и катализируют активацию различных нисходящих сигнальных молекул, тем самым способствуя восстановлению повреждений ДНК [30–32]. Кроме того, восприимчивость клеток глиобластомы к лучевой терапии и выбранный процесс репарации ДНК изменяются с активацией ряда онкогенов (например, белка 6 В-клеточной лимфомы (BCL6) и рецептора эпидермального фактора роста варианта III (EGFRvIII)) или инактивацией онкосупрессоров (например, опухолевыого супрессора p53-связывающего белок 1 (53BP1)), участвующих в повреждении и репарации ДНК, транслокациях, взаимодействиях и взаимной регуляции [33][34]. Важным исследовательским методом для повышения эффективности терапии глиобластомы является нацеливание на ключевые регуляторы в пути DDR и снижение толерантности опухолевых клеток к лучевой терапии путем нарушения регуляторной системы DDR.
Глиобластома демонстрирует значительную фенотипическую, морфологическую и клеточную гетерогенность и, как полагают, содержит популяцию самообновляющихся опухолевых стволовых клеток (ОСК), которые способствуют возникновению резистентности к терапии и склонности глиобластом к рецидивированию [35]. Одним из объяснений резистентности, опосредованных влиянием ОСК, является высокий уровень стресса репликации ДНК, вызванного воздействием радиации, который активирует DDR [36]. ОСК постоянно демонстрируют стрессовую репликацию, вызванную столкновениями репликации/транскрипции и последующей активацией DDR, что запускает резистентность к лучевой терапии. Известно, что ОСК глиобластом функционально охарактеризованы на основе их экспрессии маркера клеточной поверхности кластера дифференцировки 133 (CD133) [37]. Ряд исследований показали, что клетки CD133+ демонстрируют значительно повышенную устойчивость к стандартным методам терапии. Кроме того, эктопическая сверхэкспрессия CD133 стимулирует способность к самообновлению и пролиферации. Учитывая бесспорную — хотя и не исключительную — роль клеток CD133+ в самообновлении и устойчивости к терапии, можно предположить, что нацеливание на ОСК глиобластом через CD133 может быть многообещающей стратегией [38]. Предыдущие исследования обнаружили связь между резистентностью к лучевой терапии и статусом CD133, где результаты показали, что популяции клеток CD133+ увеличивают базальный ответ на DSB, демонстрируя активное фосфорилирование белков, связанных с контрольными точками клеточного цикла, такими как Rad17, киназа контрольной точки 1 (CHK1) и киназа контрольной точки 2 (CHK2) [39][40].
Как уже было сказано выше, TMZ и облучение являются важнейшими компонентами современной мультимодальной стандартной терапии глиобластом. Воздействие ионизирующего излучения на опухолевую клетку вызывает необратимое кластерное повреждение ДНК, а именно межцепочечные сшивки (SSB и DSB); в то время как влияние TMZ вызывает несоответствие пар оснований. Таким образом, TMZ и ионизирующее излучение действуют, повреждая ДНК, и используются для запуска гибели клеток [41]. Однако после терапии первичной глиобластомы очень часто неизбежно происходит рецидив, и это во многом связано с резистентными ОСК. Ранние исследования показали выраженную резистентность ОСК к химиотерапевтическим агентам, включая TMZ. ОСК глиобластомы демонстрируют эффективные системы восстановления повреждений ДНК, поскольку клетки CD133+ демонстрируют повышенную экспрессию MGMT, псевдогена 1 кластера точек разрыва (BCRP1) и антиапоптотических белков, которые способствуют сильному повышению устойчивости клеток CD133+ к TMZ по сравнению с их аналогами CD133–. В то время как MGMT-отрицательные ОСК оказались чувствительными к лечению TMZ, MGMT-экспрессирующие ОСК были довольно устойчивы, поскольку TMZ не мог блокировать их способность к самообновлению [42][43].
Отсутствие уникального и однозначного маркера ОСК глиобластом не позволяет прийти к окончательному ответу об эффективности алкилирующих препаратов. В ОСК глиобластом существует сложное взаимодействие между активацией сигнализации PI3K/Akt, потерей активности PTEN и резистентностью к терапии (рис. 3).
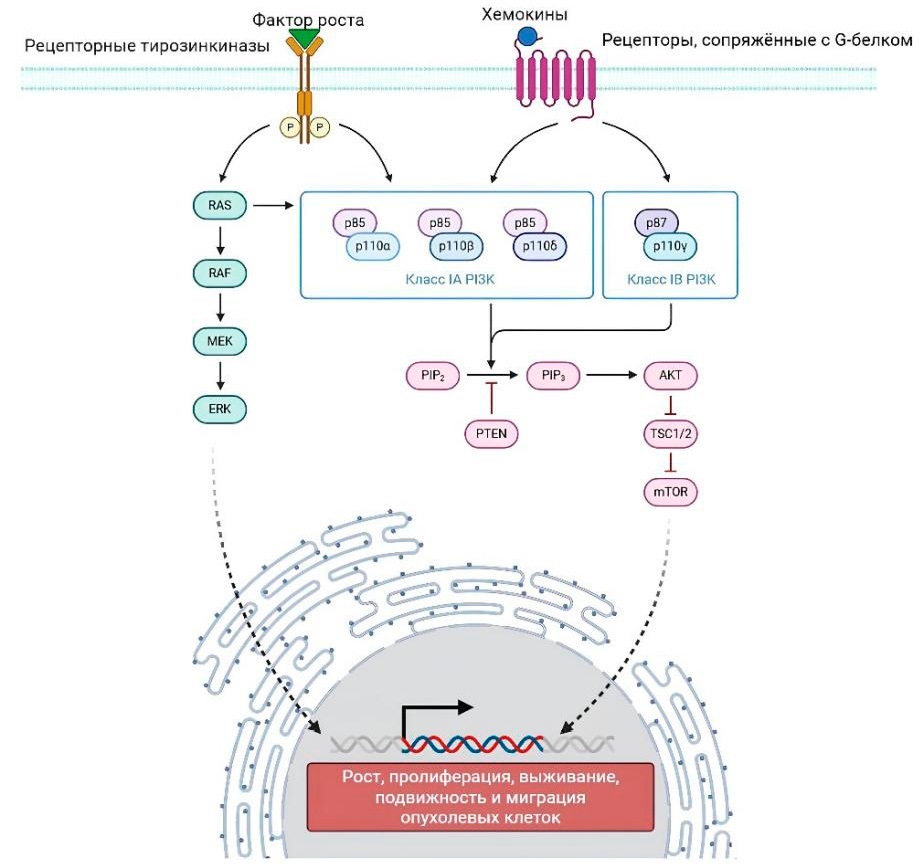
Рисунок 3. Сигнальный путь фосфоинозитид-3-киназа (PI3K)/Akt при глиобластоме. Изоформы PI3K класса I представляют собой гетеродимеры, состоящие из субъединиц p110 и p85 или p87/p101. PI3K класса IA могут активироваться рецепторными тирозинкиназами, рецепторами, сопряженными с G-белком, RAS и другими адаптерными белками, в то время как PI3K класса IB активируется исключительно рецепторами, сопряженными с G-белком. Когда PI3K активируется сигналами восходящего потока, фосфатидилинозитол 3,4,5-бисфосфат генерируется из фосфатидилинозитол 4,5-бисфосфата и активирует сигнальные пути нисходящего потока, такие как путь Akt/мишень рапамицина у млекопитающих (mTOR). Активированный путь PI3K в конечном счете способствует росту опухолевых клеток, пролиферации, выживанию, подвижности и миграции
Figure 3. PI3K/Akt Signaling Pathway in Glioblastoma. Class I Phosphoinositide 3-kinases (PI3Ks) isoforms are heterodimers consisting of p110 and p85 or p87/p101 subunits. Class IA PI3K can be activated by receptor tyrosine kinases, G protein-coupled receptors, RAS, and other adaptor proteins, while class IB PI3K is exclusively activated by G protein-coupled receptors. Upon upstream signal activation, PI3K generates phosphatidylinositol 3,4,5-trisphosphate from phosphatidylinositol 4,5-bisphosphate, triggering downstream pathways such as Akt/mammalian target of rapamycin (mTOR). Ultimately, the activated PI3K pathway promotes tumor cell growth, proliferation, survival, motility, and migration
Ингибиторы Akt или индукция экспрессии PTEN могут обратить резистентность и сенсибилизировать ОСК глиобластом к химио- и лучевой терапии, нарушая пути репарации повреждений ДНК.
Репарация ДНК и метаболические пути жизненно важны для поддержания клеточного гомеостаза в нормальных клетках человека. Однако оба эти пути претерпевают значительные изменения во время онкогенеза, включая модификации, способствующие быстрому росту, генетической гетерогенности и выживанию. Хотя эти две области исследований остаются относительно разными, появляется все больше доказательств того, что эти пути взаимозависимы и неразрывно связаны. Терапевтические вмешательства, нацеленные на метаболизм или системы репарации ДНК, вошли в клиническую практику в последние годы, подчеркивая потенциал нацеливания на эти пути при некоторых типах опухолей [44][45]. Высокое потребление глюкозы является общей характеристикой большинства солидных опухолей, и это явление было впервые описано в 1920 году Отто Варбургом. Это наблюдение, называемое эффектом Варбурга, описывает, как опухолевые клетки переключают свой преобладающий метаболический путь с окислительного фосфорилирования на анаэробный гликолиз, в результате чего вырабатывается большое количество молочной кислоты посредством ферментации (рис. 4) [46].
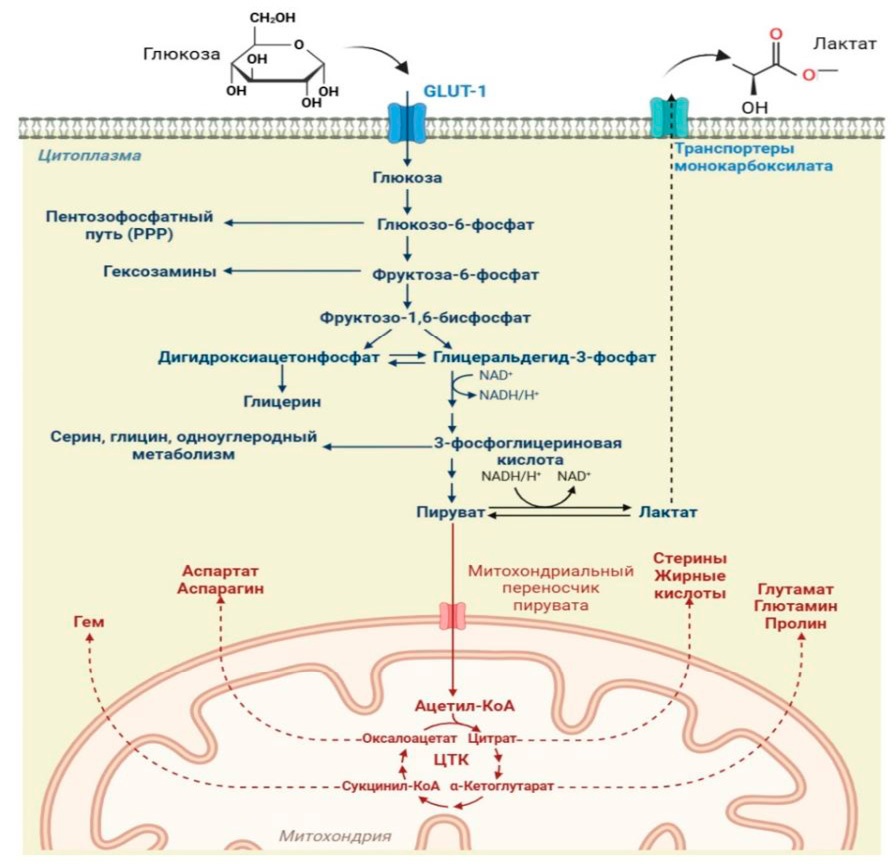
Рисунок 4. Эффект Варбурга
Figure 4. Warburg effect
Недавние исследования показали, что повышенное производство молочной кислоты может вызывать устойчивость к основным методам противоопухолевой терапии, включая химио- и лучевую терапию, посредством многочисленных механизмов [47][48]. Кроме того, повышенное производство молочной кислоты способствует развитию кислой микросреды опухоли, что связано с повышенной метастатической способностью и скоростью роста в подгруппе агрессивных опухолей, в том числе глиобластом. В опухолевых клетках, которые подвергаются метаболическому перепрограммированию, наблюдается заметное увеличение активации путей репарации на повреждение ДНК, которые впоследствии запускают синтез нуклеотидов и анаболический метаболизм глюкозы. Пути ответа на повреждение ДНК очень активны в опухолевых клетках, впоследствии способствуя их быстрому росту и выживанию. Реакция на повреждение ДНК состоит из нескольких путей репарации ДНК, и каждый путь представляет собой определенный механизм для восстановления определенного типа повреждения ДНК. Инициирование и прогрессирование путей репарации ДНК считается пространственно-временным регулируемым процессом, в котором белки перемещаются к участкам повреждения ДНК после ремоделирования хроматина [44][45]. С точки зрения химио- и лучевой терапии, восстановление DSB через NHEJ и HRR является важным фактором, поскольку многие методы лечения, включая лучевую терапию, ингибиторы топоизомеразы, такие как доксорубицин (DOX), и ингибиторы поли (АДФ-рибоза) полимеразы (PARP), индуцируют DSB ДНК [49]. Следовательно, дефектное функционирование путей восстановления DSB может существенно влиять на реакцию опухоли на эти методы лечения. Например, снижение экспрессии белков BRCA1 и BRCA2 может приводить к дефектам в HRR DSB ДНК, повышая чувствительность опухолевых клеток к ингибиторам PARP и лучевой терапии, которые вызывают повреждения, требующие HRR для восстановления [50].
Результаты некоторых исследований показали, что вероятность развития резистентности к лучевой терапии зависит от нескольких факторов, включая метаболические изменения и повышение активности путей репарации ДНК [51][52]. Метаболическое перепрограммирование может позволить опухолевым клеткам усилить синтез нуклеотидов посредством повышения активности пентозофосфатного пути (PPP), что впоследствии повышает устойчивость к традиционным методам лечения опухолей [53]. В поддержку этого ряд исследований показали, что повышение активности метаболических ферментов или метаболических процессов повышает активность путей репарации ДНК. Например, в результате повышенной гликолитической активности некоторые опухоли генерируют высокий уровень лактата, который может способствовать устойчивости к цисплатину посредством повышения активности репарации ДНК [54][55]. Как обсуждалось ранее, несколько метаболических ферментов гликолиза и PPP играют прямую роль в путях репарации ДНК, и ингибирование ключевых ферментов обоих путей не только подавляло клеточную пролиферацию, но и восстанавливало чувствительность к лучевой терапии за счет снижения активности репарации ДНК. Связь между резистентностью к лучевой терапии и измененным метаболизмом в глиобластоме до конца не изучена, но результаты некоторых исследований демонстрируют, что снижение метаболической активности ключевых ферментов, участвующих в путях PPP и гликолиза, может восстановить чувствительность некоторых резистентных опухолей к традиционным методам лечения [56–58].
При глиобластоме два гликолитических фермента, гексокиназа 2 (HK2) и изоформа M2 пируваткиназы (PKM2), были предложены в качестве перспективных целей из-за их положительной корреляции с химио- и лучевой резистентностью через антиапоптотические и клеточные механизмы выживания [59]. Например, существует четыре изоформы пируваткиназы; однако изоформа PKM2 является ключевым регулятором гликолиза в опухолевых клетках и, таким образом, является наиболее потенциальным кандидатом для восстановления чувствительности к терапии [59]. Подтверждая это, ингибирование PKM2 в клетках глиобластомы приводит к снижению жизнеспособности клеток, остановке клеточного цикла G2/M и способствует апоптозу [60]. Кроме того, ингибирование PKM2 может вызывать чувствительность к лучевой терапии, как продемонстрировано в исследовании, которое показало, что инактивация PKM2 снижает фосфорилирование Akt и киназы пируватдегидрогеназы 1 (PDK1), что впоследствии способствует чувствительности к лучевой терапии [61].
L-лактат вырабатывается в результате гликолиза и, как было обнаружено, экспрессируется в глиобластоме. Высокий уровень лактата также был связан с устойчивостью к химиотерапии у пациентов с глиобластомой [62]. Недавние исследования показали, что лактат может ингибировать активность гистондеацетилаз (HDAC), что приводит к изменениям в структуре хроматина и транскрипции. HDAC удаляют ацетильные группы из гистонов, и их ингибирование приводит к увеличению ацетилирования гистонов, которые обычно связаны с более открытой структурой хроматина для содействия транскрипции. Также предполагается, что это открытое состояние хроматина увеличивает доступность белков репарации ДНК к участкам повреждения, в свою очередь увеличивая скорость репарации ДНК [63]. Таким образом, характерное увеличение уровня лактата в клетках глиобластомы приводит к повышению активности репарации ДНК. Лактатдегидрогеназа (LDHA) — ключевой метаболический белок, обнаруженный почти во всех тканях человека, который необходим для превращения пирувата в молочную кислоту, играя важную роль на последних этапах гликолиза. Повышенная экспрессия LDHA вызывает гипоксическую среду, которая связана с метастазами, плохой общей выживаемостью и резистентностью к химиолучевой терапии у пациентов с глиобластомой [64][65]. На основании этих результатов можно предположить, что ингибирование активности LDHA может придавать клеткам глиобластомы чувствительность к агентам, повреждающим ДНК. Дальнейшее изучение связей между метаболическими и репарационными путями ДНК может открыть новые терапевтические подходы против глиобластомы в будущем.
Важность путей DDR в поддержании жизнеспособности клеток и предотвращении неоплазии подчеркивается дополнительными неотъемлемыми ролями этих путей в регуляции клеточного цикла, ремоделирования хроматина, метаболизма, иммуногенности и апоптоза. Например, обнаружение повреждения ДНК приводит к активации контрольных точек, которые обеспечивают остановку клеточного цикла, чтобы предоставить время, необходимое для восстановления ДНК перед делением клетки; пути DDR также тесно связаны с апоптотическим механизмом, чтобы обеспечить устранение клеток с невосстановленным повреждением ДНК (рис. 5) [66–68].
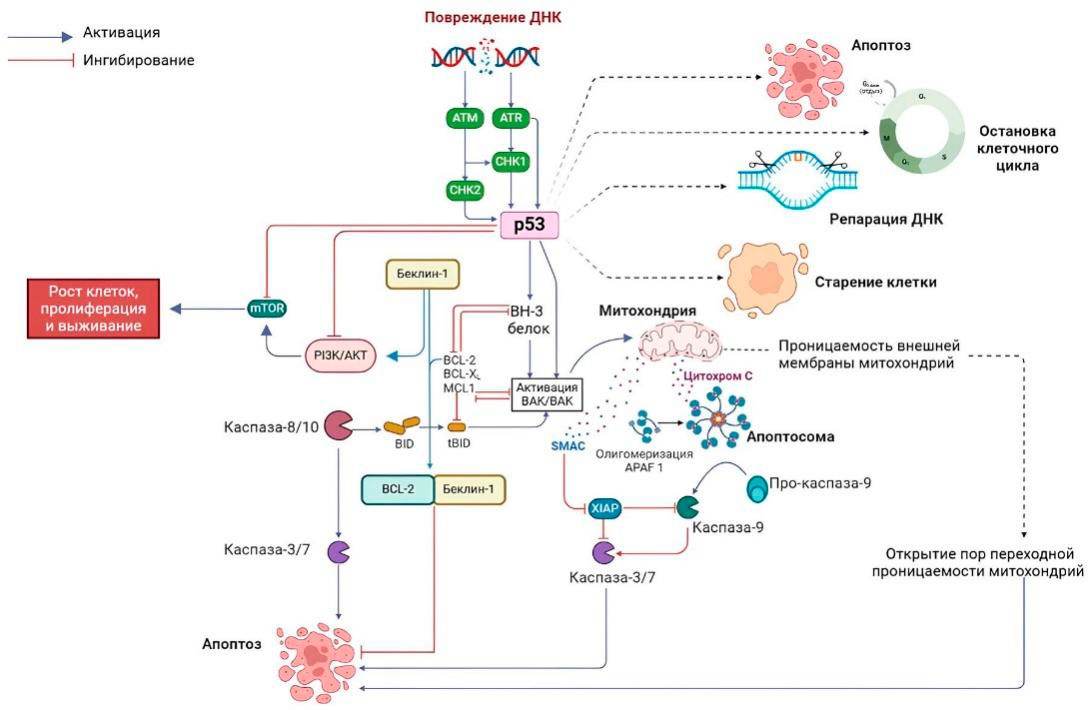
Рисунок 5. Повреждение ДНК и апоптоз. P53 является главным регулятором клеточной смерти и активируется через различные клеточные процессы, например в ответ на повреждение ДНК. Активация p53 приводит к апоптозу. Кроме того, апоптоз может активироваться в зависимости от состояния клетки. Потенциал внешней мембраны митохондрий нарушается при активации внутреннего пути апоптоза, что приводит к открытию пор переходной проницаемости митохондрий. В этой ситуации количество пораженных митохондрий определяет судьбу клетки. Если поражено только несколько митохондрий, активность аутофагии может устранить их, что приводит к выживанию клетки. Однако когда поражено больше митохондрий, клеточной судьбой будет апоптотическая смерть
Figure 5. DNA damage and apoptosis. The p53 protein is a primary regulator of programmed cell death (apoptosis) and can be activated in response to various cellular processes, such as DNA damage. p53 activation induces apoptosis, which may also be triggered depending on cellular state. The activation of intrinsic apoptosis involves a decrease in mitochondrial outer membrane potential, which in turn leads to mitochondrial permeability transition pore opening. The extent of mitochondrial damage dictates cell fate: if only a few are affected, autophagy might eliminate them, promoting cell survival. However, extensive mitochondrial damage results in apoptotic death
Таким образом, пути DDR в конечном счете обеспечивают выживание клеток в условиях геномной нестабильности и репликативного стресса или направляют непоправимо поврежденные клетки на старение или запрограммированную смерть. Геномная нестабильность является ключевым признаком любой опухоли и возникает в результате высокой скорости деления клеток и связанного с этим быстрого накопления аберраций на фоне нарушенных процессов DDR, которые способствуют возникновению и прогрессированию опухоли. Следовательно, дефекты в генах DDR играют множественную роль в содействии роста опухоли посредством накопления драйверных мутаций, генерации гетерогенности опухоли и уклонения от апоптоза [66]. Как было сказано выше, существует противоопухолевая терапия посредством использования ДНК-повреждающей лучевой терапии и ряда химиопрепаратов при глиобластоме, а в последнее время идет разработка мощных и селективных молекулярно-таргетных агентов против ключевых компонентов различных путей DDR (далее именуемых ингибиторами DDR). Однако разработка аналитически и клинически подтвержденных анализов для надежной оценки прогностических биомаркеров ответа и/или устойчивости к ингибиторам DDR отстает.
PARP-ингибиторы
Ингибиторы PARP недавно были исследованы в качестве сенсибилизирующих препаратов для усиления эффективности TMZ. PARP — это класс ферментов, который участвует в пути BER, а также в пути MGMT, физически взаимодействуя с MGMT и в конечном итоге PARилирует в ответ на химиотерапию TMZ для устранения аддуктов O6-MG из поврежденного сегмента ДНК. Во-вторых, PARP работает как сенсор, запуская пути ответа BER. Препараты — ингибиторы PARP блокируют связывание PARP-MGMT или PARилируют MGMT, снижая функцию MGMT и предотвращая восстановление O6-MG. В результате функция MGMT снижается, что приводит к сенсибилизации TMZ и дает обоснование для сенсибилизации [69][70].
Препараты — ингибиторы PARP исследовались у пациентов с глиобластомой в ряде клинических испытаний. Однако множество факторов затрудняют клиническую разработку этих ингибиторов. Ингибиторы PARP могут вызывать синтетическую летальность в опухолевых клетках с существующими дефектами в путях репарации HRR, таких как вредные мутации генов — супрессоров BRCA1 и BRCA2. BRCA1 и BRCA2 играют роль в восстановлении ДНК и необходимы для стабильности генома. Более того, поскольку наличие функционального белка BRCA1 и BRCA2, по-видимому, является предиктивным биомаркером неблагоприятного исхода выживания пациентов с глиобластомами, возможно, что агенты, которые подавляют экспрессию BRCA1 и BRCA2, могут быть использованы в качестве новой терапевтической стратегии для пациентов с глиобластомой с нормальным или высоким уровнем белка BRCA1/2, сенсибилизируя их к лечению, повреждающему ДНК [71–73]. Проще говоря, нацеливание на BRCA1/2 может модулировать восстановление ДНК и потенциально повышать эффективность лучевой терапии и алкилирующих агентов у пациентов с глиобластомой (рис. 6).
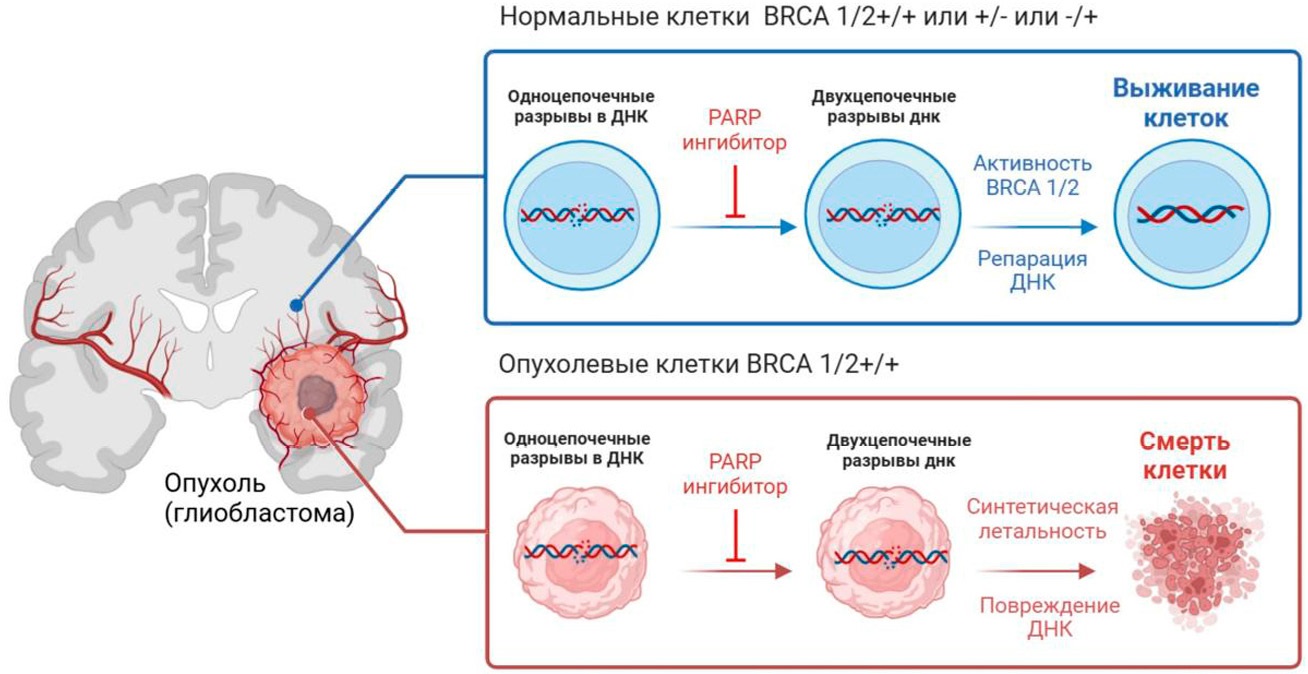
Рисунок 6. Потенциал использования ингибиторов поли (АДФ-рибоза) полимеразы (PARP) у пациентов с глиобластомами и вредными мутациями гена рака молочной железы 1 (BRCA1) и гена рака молочной железы 1 (BRCA2)
Figure 6. Therapeutic potential of poly (ADP-ribose) polymerase (PARP) inhibitors for glioblastoma patients harboring deleterious breast cancer gene 1 (BRCA1) or breast cancer gene 2 (BRCA2) mutations
Примеры активно действующих ингибиторов PARP против глиобластомы с их доклинической и клинической значимостью приведены в таблице 1 [74–83] и таблице 2 (https://clinicaltrials.gov/).
Препарат | Описание | Литература |
Рукапариб | Показано, что препарат эффективен в сенсибилизации клеток глиобластом, резистентных к иринотекану и TMZ без усиления миелотоксических эффектов in vivo. Сообщается о полной регрессии опухоли в течение более 60 дней при применении в сочетании с TMZ in vivo | [74][75] |
Олапариб | Увеличение чувствительности к лучевой терапии глиобластом, эпендимом и медуллобластомы | [76] |
Нирапариб | При одновременном применении с лучевой терапией наблюдалось постепенное улучшение показателей выживаемости мышей в моделях глиобластомы и диффузной внутренней глиомы моста | [77] |
Талазопариб | Сообщается о чувствительности к EFGR-амплифицированным клеткам глиобластомы, формирующим нейросферы. В опухолях с дефицитом репарации ДНК наблюдалось синергическое действие с препаратами на основе платины и TMZ | [78][79] |
Велипариб | Сообщается о высокой эффективности в сочетании с TMZ против клеток глиобластомы с дефицитом PTEN in vivo. Также обнаружено, что препарат эффективен при лечении моделей ксенотрансплантатов, полученных от пациентов, и клеточных линий глиобластом с неметилированным MGMT при применении вместе с лучевой терапией | [80–82] |
Памипариб | В исследованиях in vivo продемонстрирована отличная проницаемость через ГЭБ и улучшение времени выживания при сочетании с TMZ | [83] |
Таблица 1. Преклинические исследования использования ингибиторов поли (АДФ-рибоза) полимеразы (PARP) в терапии глиобластомы
Table 1. Preclinical studies of PARP inhibitors in glioblastoma treatment
Препарат | Фаза | Описание | Номер клинического исследования |
Памипариб | Фаза 2 | Исследовали эффективность препарата у пациентов с впервые диагностированной глиобластомой (неметилированный промотор MGMT) и у пациентов с рецидивирующей глиобластомой | NCT04614909 |
Олапариб | Фаза 2 | Определить безопасность и эффективность олапариба и TMZ в комбинации; посмотреть, насколько хорошо эти препараты работают при совместном применении у людей с глиобластомой, которые либо не реагировали на предыдущее лечение, либо произошел рецидив после лечения Целью этого исследования было посмотреть, насколько эффективен препарат у пациентов с рецидивирующей глиобластомой и мутацией IDH на основе их 6-месячной выживаемости. Оценить эффективность комбинированной терапии, сосредоточившись на средней частоте ответа и частоте профилактики заболеваний. Считается, что сочетание дурвалумаба и олапариба будет более полезным для пациентов с мутацией IDH с солидными опухолями, чем каждый из препаратов по отдельности В этом исследовании изучается эффективность препарата при лечении холангиокарциномы и глиом, которые имеют мутацию в гене IDH1 или IDH2 (не поддаются лечению или контролю с помощью современных методов лечения) | NCT05188508 NCT03561870 NCT03991832 |
Нирапариб | Фаза 2 | Испытание проводилось для оценки эффективности, а также безопасности препарата с полями для лечения опухолей (TTFields) у пациентов с рецидивирующей глиобластомой Изучалоась безопасность и эффективность препарата в сочетании с лучевой терапией у лиц с рецидивирующей глиобластомой | NCT04221503 NCT04715620 |
Велипариб | Фаза 2 | Изучался комбинированный эффект препарата и TMZ, а также лучевой терапии при недавно выявленной глиобластоме без мутаций в BRAFV600 или H3K27M Сравнение эффективности препарата и TMZ в комбинации и по отдельности при лечении недавно диагностированной глиобластомы | NCT03581292 NCT02152982 |
Таблица 2. Клинические исследования использования ингибиторов поли (АДФ-рибоза) полимеразы (PARP) в терапии глиобластомы
Table 2. Clinical trials of PARP inhibitors in glioblastoma therapy
Ингибиторы ATM
ATM, незаменимая киназа, регулирующая HRR, повсеместно экспрессируется в опухолевых клетках [84]. ATM является перспективной терапевтической мишенью, поскольку ее ингибирование, вероятно, сенсибилизирует опухоли к повреждающему ДНК эффекту лучевой терапии и ряда химиопрепаратов [85]. MMR преобразует аддукты ДНК, индуцированные TMZ, во вторичные поражения, которые блокируют репликативную вилку, тем самым приводя к DSB и активации ферментов DDR [14][16]. Было показано, что TMZ активирует сигнальные пути, зависимые от ATM [85]. Например, было продемонстрировано, что в клетках глиобластомы, обладающих MMR, воздействие низкой дозы TMZ активирует ATM и приводит к фосфорилированию CHK1, CHK2 и p53 и остановке клеточного цикла G2/M [86].
Существуют доклинические исследования, доказывающие роль p53 как биомаркера ответа на ингибирование ATM при глиобластоме. Например, генетическая инактивация кофактора ATM (ATMIN) подавляет образование глиобластомы in vivo с дефицитом p53 [87]. Кроме того, сочетание ингибиторов ATM и ингибиторов рецептора фактора роста тромбоцитов альфа (PDGFRA) снижает выживаемость клеток глиобластомы с мутацией p53, что указывает на роль ингибиторов ATM в лечении пациентов с глиобластомой с мутациями p53 [88]. Аналогично, использование KU60019, аналога ингибитора ATM второго поколения, приводит к более выраженной чувствительности к лучевой терапии в клеточной линии глиобластомы U87 с мутацией p53, чем в генетически соответствующих клетках дикого типа [89]. Кроме того, эффективность KU60019 была связана с ингибированием фосфорилирования основных эффекторов повреждения ДНК p53, H2AX, KAP1 и Akt. Известно, что KU60019 необычайно стабилен, но не может пересекать ГЭБ.
Ингибиторы ATM нового поколения, такие как AZ32 и AZD1390 (AstraZeneca), специально разработаны для пересечения ГЭБ [90]. По сравнению с лучевой терапией в отдельности, комбинация AZD1390 и лучевой терапии вызывает значительную регрессию глиобластомы [91]. AZD1390, наиболее клинически продвинутый ингибитор ATM для лечения глиобластомы и метастатических опухолей в головной мозг, проходит испытания в фазе I (NCT03423628) (https://clinicaltrials.gov/). Испытание оценивает безопасность и переносимость AZD1390 в сочетании с модулированной по интенсивности лучевой терапией у пациентов с рецидивирующей глиобластомой (35 Гр в течение 2 недель) или недавно диагностированной первичной глиобластомой (60 Гр в течение 6 недель) и в сочетании с полной или частичной лучевой терапией мозга (30 Гр в течение 2 недель) у пациентов с метастазами в головной мозг.
Ингибиторы ATR
Сигнальный путь ATR-CHK1, основной эффектор контрольных точек репликации и повреждения ДНК, предотвращает вступление клеток с поврежденной ДНК в митоз [85]. ATR представляет особый интерес в терапии глиобластомы, поскольку он играет доминирующую роль в защите опухолевых клеток от TMZ [92]. Как и в случае с другими ингибиторами DDR, существуют некоторые опасения относительно токсичности ингибиторов ATR для нормальных клеток, поскольку ATR необходим для выживания многих типов клеток. Снижение ATR усиливает апоптоз, вызванный TMZ, в клетках глиобластомы. Помимо индукции апоптоза, TMZ также активирует пути выживания, такие как старение. Отличительными признаками клеточного старения являются активация DDR и остановка клеточного цикла, что позволяет клеткам выживать без пролиферации и способствует рецидиву. Старение, вызванное TMZ, в клетках глиобластомы зависит от активации сигнального пути ATR-CHK1 [93][94]. Имеются доказательства того, TMZ активирует ATR зависимым от MGMT образом и что использование TMZ в клетках глиобластомы с дефицитом MGMT увеличивает чувствительность к ингибиторам ATR в моделях глиобластомы in vitro и in vivo [95].
Насколько нам известно, в настоящее время не проводятся клинические испытания ингибиторов ATR для глиобластомы (www.clinicaltrials.org). M6220 также изучается в сочетании с лучевой терапией у пациентов с немелкоклеточным раком легких с метастазами в головной мозг (NCT02589522). Новый мощный селективный ингибитор ATR, Элимусертиб (BAY1895344), по-видимому, имеет приемлемый профиль безопасности в качестве монотерапии у пациентов с прогрессирующими солидными опухолями.
Помимо прямого комбинированного воздействия на факторы DDR, сопоставимый подход может заключаться в воздействии на другие сигнальные пути, которые влияют на активность и/или емкость DDR. Например, было установлено, что нарушение функциональных сигнальных путей фактора роста эндотелия сосудов (VEGF) и Akt влияет на баланс между NHEJ и активностью восстановления разрывов ДНК HR в ОСК глиобластом, что приводит к повышению чувствительности к лучевой терапии [96]. Это особенно интересно, учитывая, что таргетная терапия против VEGF с помощью бевацизумаба в целом не смогла улучшить общую выживаемость пациентов с глиобластомой в крупных клинических испытаниях [97]. Аналогично выявлению неклассических стратегий нацеливания на пути DDR недавно был идентифицирован с помощью киномного скрининга РНК-интерференции митоген-активируемая протеинкиназа 7 (ERK5)/киназа-активируемая протеинкиназа 5 (MAPK5) сигнальный путь как новый фактор устойчивости к TMZ с инактивацией ERK5 в клетках глиобластомы, что в итоге привело к дефектной способности к восстановлению ДНК, вероятно, из-за ненадлежащей активности NHEJ перед митозом [98]. Интересно, что ERK5 недавно был идентифицирован как ключевой фактор в содействии росту клеток и выживанию клеток в агрессивных диффузных внутренних понтинных глиомах, что подтверждает недавние данные о ERK5 как о новой терапевтической мишени [99].
Недавние открытия выявили ключевые молекулярные и функциональные связи между DDR, сигнализацией репликационного стресса и иммунными путями циклической ГМФ-АМФ-синтаза (cGAS)-стимулятор генов интерферона (STING), а также то, что нацеливание на сигнализацию репликационного стресса может синергировать с иммунотерапией глиобластом (рис. 7) [100–102].
После открытия того, что cGAS-STING распознает эндогенную ДНК, высвобождаемую из умирающих опухолевых клеток, и индуцирует интерферон I типа и противоопухолевые реакции Т-клеток, были предприняты попытки понять и терапевтически воздействовать на путь STING при опухолях. По сравнению с другими типами злокачественных новообразований иммунная микросреда глиобластомы содержит мало инфильтрирующих Т-клеток, но большое количество миелоидных клеток, ассоциированных с опухолью, что, возможно, объясняет неутешительные ответы на терапию блокадой иммунных контрольных точек у групп пациентов с глиобластомой. Примечательно, что в отличие от большинства экстракраниальных опухолей экспрессия STING отсутствует в глиобластоме, вероятно, из-за метилирования промотора STING. Тем не менее несколько доклинических исследований показывают, что индуцирование cGAS-STING сигнализации в иммунной микросреде глиобластомы может быть терапевтически полезным, и было показано, что cGAS-STING сигнализация опосредует воспалительные и противоопухолевые эффекты других модальностей, которые либо используются, либо разрабатываются для терапии глиобластомы, включая лучевую терапию и онколитическую виротерапию.
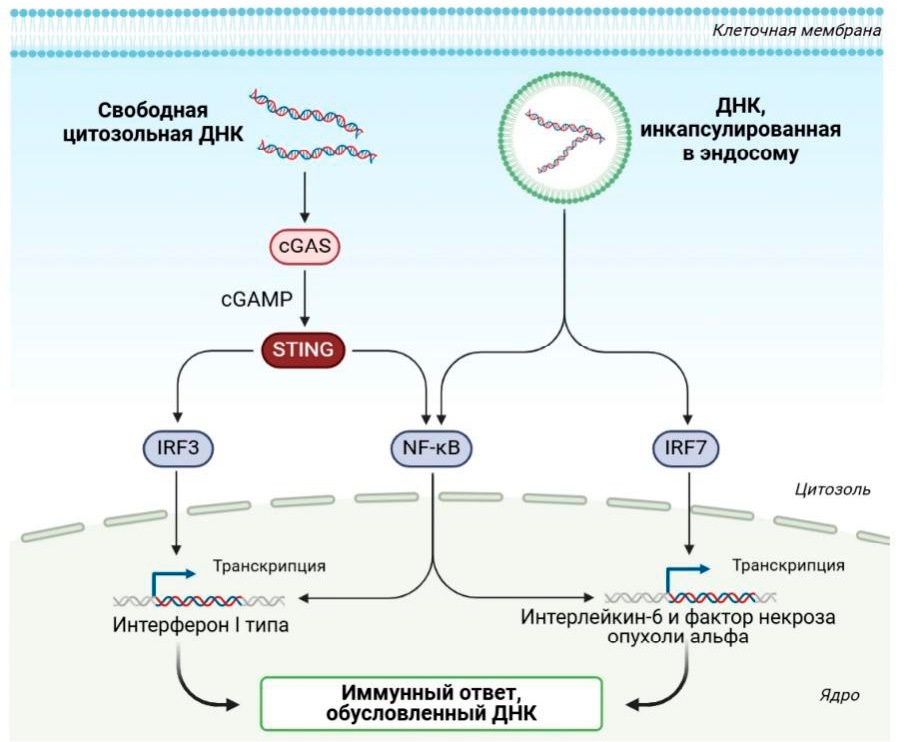
Рисунок 7. Циклическая ГМФ-АМФ-синтаза (cGAS)-стимулятор генов интерферона (STING) ДНК-детекция
Figure 7. Cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes (STING) DNA detection
Способность клеток восстанавливать ДНК после повреждений из эндогенных или экзогенных источников имеет важное значение для поддержания их нормальной жизнеспособности. Различные виды повреждений возникают из разных источников и требуют определенных путей репарации повреждений ДНК, позволяющих восстановить исходную последовательность. Повреждения, оставшиеся невосстановленными, могут быть унаследованы после деления клетки, вызывая постоянные генетические изменения. Накопление этих мутаций приводит к старению клеток или апоптозу и может предрасполагать к развитию опухолей, в том числе глиобластом. Более того, в процессе старения способность клеток к восстановлению ДНК снижается, а также претерпевает метаболические изменения, вызванные клеточными и эндокринными изменениями. В случае глиобластомы дальнейшие исследования могут также выявить новые терапевтические мишени, которые могут быть нацелены как на метаболизм, так и на восстановление ДНК одновременно.
Нестабильность генома клеток глиобластом возникает из-за различных дефектов в механизме репарации ДНК, которые делают их более восприимчивыми к агентам, воздействующим на ДНК. Взаимосвязь между дефицитом репарации ДНК и повышенным эффектом агентов, воздействующих на ДНК, подчеркивает DSB, которая включает пути HRR и NHEJ. Препараты, воздействующие на ДНК, являются многообещающими терапевтическими средствами с точным применением на фоне специфической для опухоли неудачи репарации ДНК. Исследование и понимание механизмов химиолучевой резистентности в клетках глиобластом имеет фундаментальное значение для разработки новых эффективных стратегий лечения, поскольку модуляция способности репарации ДНК может быть средством повышения клеточной чувствительности к генотоксическим агентам. Поэтому контролируемое целевое ингибирование факторов DDR в сочетании с химиотерапевтическими препаратами будет представлять собой полезную стратегию для предотвращения временной остановки клеточного цикла и восстановления повреждений ДНК, для содействия гибели опухолевых клеток и улучшения результатов лечения пациентов с глиобластомой.
1. Roda D., Veiga P., Melo J.B., Carreira I.M., Ribeiro I.P. Principles in the Management of Glioblastoma. Genes (Basel). 2024;15(4):501. DOI: 10.3390/genes15040501
2. Read R.D., Tapp Z.M., Rajappa P., Hambardzumyan D. Glioblastoma microenvironment-from biology to therapy. Genes Dev. 2024;38(9– 10):360–79. DOI: 10.1101/gad.351427.123
3. Németh E., Szüts D. The mutagenic consequences of defective DNA repair. DNA Repair (Amst). 2024;139:103694. DOI: 10.1016/j.dnarep.2024.103694
4. Hopkins J.L., Lan L., Zou L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022;36(5–6):278–93. DOI: 10.1101/gad.349431.122
5. Ikliptikawati D.K., Hirai N., Makiyama K., Sabit H., Kinoshita M., Matsumoto K., et al. Nuclear transport surveillance of p53 by nuclear pores in glioblastoma. Cell Rep. 2023;42(8):112882. DOI: 10.1016/j.celrep.2023.112882
6. Le Rhun E., Preusser M., Roth P., Reardon D.A., van den Bent M., Wen P., et al. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. 2019;80:101896. DOI: 10.1016/j.ctrv.2019.101896
7. Butler M., Pongor L., Su Y.T., Xi L., Raffeld M., Quezado M., et al. MGMT Status as a clinical biomarker in glioblastoma. Trends Cancer. 2020;6(5):380–91. DOI: 10.1016/j.trecan.2020.02.010
8. Hashemi M., Etemad S., Rezaei S., Ziaolhagh S., Rajabi R., Rahmanian P., et al. Progress in targeting PTEN/PI3K/Akt axis in glioblastoma therapy: Revisiting molecular interactions. Biomed Pharmacother. 2023;158:114204. DOI: 10.1016/j.biopha.2022.114204
9. Oksenych V., Kainov D.E. DNA Damage Response. Biomolecules. 2021;11(1):123. DOI: 10.3390/biom11010123
10. Chappidi N., Quail T., Doll S., Vogel L.T., Aleksandrov R., Felekyan S., et al. PARP1-DNA co-condensation drives DNA repair site assembly to prevent disjunction of broken DNA ends. Cell. 2024;187(4):945–61. e18. DOI: 10.1016/j.cell.2024.01.015
11. Cheng B., Pan W., Xing Y., Xiao Y., Chen J., Xu Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur J Med Chem. 2022;230:114109. DOI: 10.1016/j.ejmech.2022.114109
12. Chatterjee N., Walker G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235–63. DOI: 10.1002/em.22087
13. Jurkovicova D., Neophytou C.M., Gašparović A.Č., Gonçalves A.C. DNA Damage Response in Cancer Therapy and Resistance: Challenges and Opportunities. Int J Mol Sci. 2022;23(23):14672. DOI: 10.3390/ijms232314672
14. Alghoul E., Basbous J., Constantinou A. Compartmentalization of the DNA damage response: Mechanisms and functions. DNA Repair (Amst). 2023;128:103524. DOI: 10.1016/j.dnarep.2023.103524
15. Vernì F. DNA damage response (DDR) and DNA repair. Int J Mol Sci. 2022;23(13):7204. DOI: 10.3390/ijms23137204
16. Wu L., Sowers J.R., Zhang Y., Ren J. Targeting DNA damage response in cardiovascular diseases: from pathophysiology to therapeutic implications. Cardiovasc Res. 2023;119(3):691–709. DOI: 10.1093/cvr/cvac080
17. Sareen H., Ma Y., Becker T.M., Roberts T.L., de Souza P., Powter B. Molecular Biomarkers in Glioblastoma: A Systematic Review and Meta-Analysis. Int J Mol Sci. 2022;23(16):8835. DOI: 10.3390/ijms23168835
18. Lee Y.C., Lee Y.L., Li C.Y. BRCA Genes and Related Cancers: A Meta-Analysis from Epidemiological Cohort Studies. Medicina (Kaunas). 2021;57(9):905. DOI: 10.3390/medicina57090905
19. Jing X., Yang F., Shao C., Wei K., Xie M., Shen H., et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157. DOI: 10.1186/s12943-019-1089-9
20. Scanlon S.E., Glazer P.M. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst). 2015;32:180–9. DOI: 10.1016/j.dnarep.2015.04.030
21. Li M., Thorne R.F., Shi R., Zhang X.D., Li J., Li J., et al. DDIT3 directs a dual mechanism to balance glycolysis and oxidative phosphorylation during glutamine deprivation. Adv Sci (Weinh). 2021;8(11):e2003732. DOI: 10.1002/advs.202003732
22. Tran T.Q., Ishak Gabra M.B., Lowman X.H., Yang Y., Reid M.A., Pan M., et al. Glutamine deficiency induces DNA alkylation damage and sensitizes cancer cells to alkylating agents through inhibition of ALKBH enzymes. PLoS Biol. 2017;15(11):e2002810. DOI: 10.1371/journal.pbio.2002810
23. Goldstein M., Kastan M.B. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–43. DOI: 10.1146/annurev-med-081313-121208
24. Shaw R., Basu M., Karmakar S., Ghosh M.K. MGMT in TMZ-based glioma therapy: Multifaceted insights and clinical trial perspectives. Biochim Biophys Acta Mol Cell Res. 2024;1871(3):119673. DOI: 10.1016/j.bbamcr.2024.119673
25. Brawanski K.R., Sprung S., Freyschlag C.F., Hoeftberger R., Ströbel T., Haybaeck J., et al. Influence of MMR, MGMT promotor methylation and protein expression on overall and progression-free survival in primary glioblastoma patients treated with temozolomide. Int J Mol Sci. 2023;24(7):6184. DOI: 10.3390/ijms24076184
26. Fahrer J., Christmann M. DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways. Int J Mol Sci. 2023;24(5):4684. DOI: 10.3390/ijms24054684
27. Tang J.B., Svilar D., Trivedi R.N., Wang X.H., Goellner E.M., Moore B., et al. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011;13(5):471–86. DOI: 10.1093/neuonc/nor011
28. Liu J., Bi K., Yang R., Li H., Nikitaki Z., Chang L. Role of DNA damage and repair in radiation cancer therapy: a current update and a look to the future. Int J Radiat Biol. 2020;96(11):1329–38. DOI: 10.1080/09553002.2020.1807641
29. Ghosh S., Ghosh A. Activation of DNA damage response signaling in mammalian cells by ionizing radiation. Free Radic Res. 2021;55(5):581–94. DOI: 10.1080/10715762.2021.1876853
30. Carusillo A., Mussolino C. DNA damage: from threat to treatment. Cells. 2020;9(7):1665. DOI: 10.3390/cells9071665
31. Choi J.E., Chung W.H. Synthetic lethal interaction between oxidative stress response and DNA damage repair in the budding yeast and its application to targeted anticancer therapy. J Microbiol. 2019;57(1):9– 17. DOI: 10.1007/s12275-019-8475-2
32. Vitale I., Kroemer G. Spontaneous DNA damage propels tumorigenicity. Cell Res. 2017;27(6):720–1. DOI: 10.1038/cr.2017.43
33. Graziano S., Gonzalo S. Mechanisms of oncogene-induced genomic instability. Biophys Chem. 2017;225:49–57. DOI: 10.1016/j.bpc.2016.11.008
34. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. DOI: 10.1146/annurev-physiol-030212-183653
35. Yabo Y.A., Niclou S.P., Golebiewska A. Cancer cell heterogeneity and plasticity: A paradigm shift in glioblastoma. Neuro Oncol. 2022;24(5):669–82. DOI: 10.1093/neuonc/noab269
36. Li C., Qiu S., Liu X., Guo F., Zhai J., Li Z., et al. Extracellular matrixderived mechanical force governs breast cancer cell stemness and quiescence transition through integrin-DDR signaling. Signal Transduct Target Ther. 2023;8(1):247. DOI: 10.1038/s41392-023-01453-0
37. Barzegar Behrooz A., Syahir A., Ahmad S. CD133: beyond a cancer stem cell biomarker. J Drug Target. 2019;27(3):257–69. DOI: 10.1080/1061186X.2018.1479756
38. Min D.W., Kim H.P., Kim J., Wen X., Kim S., Cho Y.W., et al. Phenotype-based single cell sequencing identifies diverse genetic subclones in CD133 positive cancer stem cells. Biochem Biophys Res Commun. 2021;558:209–15. DOI: 10.1016/j.bbrc.2020.09.005
39. Ropolo M., Daga A., Griffero F., Foresta M., Casartelli G., Zunino A., et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol Cancer Res. 2009;7(3):383–92. DOI: 10.1158/1541-7786.MCR-08-0409
40. Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60. DOI: 10.1038/nature05236
41. Cantidio F.S., Gil G.O.B., Queiroz I.N., Regalin M. Glioblastoma — treatment and obstacles. Rep Pract Oncol Radiother. 2022;27(4):744– 53. DOI: 10.5603/RPOR.a2022.0076
42. Liu G., Yuan X., Zeng Z., Tunici P., Ng H., Abdulkadir I.R., et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. DOI: 10.1186/1476-4598-5-67
43. Beier D., Schulz J.B., Beier C.P. Chemoresistance of glioblastoma cancer stem cells — much more complex than expected. Mol Cancer. 2011;10:128. DOI: 10.1186/1476-4598-10-128
44. Puigvert J.C., Sanjiv K., Helleday T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016;283(2):232–45. DOI: 10.1111/febs.13574
45. Efimova E.V., Takahashi S., Shamsi N.A., Wu D., Labay E., Ulanovskaya O.A., et al. Linking cancer metabolism to DNA repair and accelerated senescence. Mol Cancer Res. 2016;14(2):173–84. DOI: 10.1158/1541-7786.MCR-15-0263
46. Barba I., Carrillo-Bosch L., Seoane J. Targeting the Warburg Effect in Cancer: Where Do We Stand? Int J Mol Sci. 2024;25(6):3142. DOI: 10.3390/ijms25063142
47. Liu X., Li Z., Zhao Q., Zhou X., Wang Y., Zhao G., et al. Capsaicin reverses cisplatin resistance in tongue squamous cell carcinoma by inhibiting the Warburg effect and facilitating mitochondrialdependent apoptosis via the AMPK/AKT/mTOR axis. Cell Biol Int. 2024;48(8):1097–110. DOI: 10.1002/cbin.12169
48. Wen S.S., Wu Y.J., Wang J.Y., Ni Z.X., Dong S., Xie X.J., et al. BRAFV600E/p-ERK/p-DRP1(Ser616) promotes tumor progression and reprogramming of glucose metabolism in papillary thyroid cancer. Thyroid. 2024;34(10):1246–59. DOI: 10.1089/thy.2023.0700
49. Pothuri B., Brodsky A.L., Sparano J.A., Blank S.V., Kim M., Hershman D.L., et al. Phase I and pharmacokinetic study of veliparib, a PARP inhibitor, and pegylated liposomal doxorubicin (PLD) in recurrent gynecologic cancer and triple negative breast cancer with long-term follow-up. Cancer Chemother Pharmacol. 2020;85(4):741–51. DOI: 10.1007/s00280-020-04030-2
50. Dibitetto D., Widmer C.A., Rottenberg S. PARPi, BRCA, and gaps: controversies and future research. Trends Cancer. 2024;10(9):857–69. DOI: 10.1016/j.trecan.2024.06.008
51. Cucchi D., Gibson A., Martin S.A. The emerging relationship between metabolism and DNA repair. Cell Cycle. 2021;20(10):943–59. DOI: 10.1080/15384101.2021.1912889
52. Koo S.Y., Park E.J., Noh H.J., Jo S.M., Ko B.K., Shin H.J., et al. Ubiquitination Links DNA Damage and Repair Signaling to Cancer Metabolism. Int J Mol Sci. 2023;24(9):8441. DOI: 10.3390/ijms24098441
53. Helleday T., Rudd S.G. Targeting the DNA damage response and repair in cancer through nucleotide metabolism. Mol Oncol. 2022;16(21):3792–810. DOI: 10.1002/1878-0261.13227
54. Govoni M., Rossi V., Di Stefano G., Manerba M. Lactate upregulates the expression of DNA repair genes, causing intrinsic resistance of cancer cells to cisplatin. Pathol Oncol Res. 2021;27:1609951. DOI: 10.3389/pore.2021.1609951
55. Fan Z., Ye M., Liu D., Zhou W., Zeng T., He S., et al. Lactate drives the ESM1-SCD1 axis to inhibit the antitumor CD8+ T-cell response by activating the Wnt/β-catenin pathway in ovarian cancer cells and inducing cisplatin resistance. Int Immunopharmacol. 2024;137:112461. DOI: 10.1016/j.intimp.2024.112461
56. Kathagen-Buhmann A., Schulte A., Weller J., Holz M., Herold-Mende C., Glass R., et al. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. 2016;18(9):1219–29. DOI: 10.1093/neuonc/now024
57. Marin-Valencia I., Cho S.K., Rakheja D., Hatanpaa K.J., Kapur P., Mashimo T., et al. Glucose metabolism via the pentose phosphate pathway, glycolysis and Krebs cycle in an orthotopic mouse model of human brain tumors. NMR Biomed. 2012;25(10):1177–86. DOI: 10.1002/nbm.2787
58. Zhu Z., Kiang K.M., Li N., Liu J., Zhang P., Jin L., et al. Folate enzyme MTHFD2 links one-carbon metabolism to unfolded protein response in glioblastoma. Cancer Lett. 2022;549:215903. DOI: 10.1016/j.canlet.2022.215903
59. Yuen C.A., Asuthkar S., Guda M.R., Tsung A.J., Velpula K.K. Cancer stem cell molecular reprogramming of the Warburg effect in glioblastomas: a new target gleaned from an old concept. CNS Oncol. 2016;5(2):101–8. DOI: 10.2217/cns-2015-0006
60. Mukherjee J., Ohba S., See W.L., Phillips J.J., Molinaro A.M., Pieper R.O. PKM2 uses control of HuR localization to regulate p27 and cell cycle progression in human glioblastoma cells. Int J Cancer. 2016;139(1):99–111. DOI: 10.1002/ijc.30041
61. Goidts V., Bageritz J., Puccio L., Nakata S., Zapatka M., Barbus S., et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene. 2012;31(27):3235–43. DOI: 10.1038/onc.2011.490
62. Khan F., Lin Y., Ali H., Pang L., Dunterman M., Hsu W.H., et al. Lactate dehydrogenase A regulates tumor-macrophage symbiosis to promote glioblastoma progression. Nat Commun. 2024;15(1):1987. DOI: 10.1038/s41467-024-46193-z
63. Nguyen T.T.T., Zhang Y., Shang E., Shu C., Torrini C., Zhao J., et al. HDAC inhibitors elicit metabolic reprogramming by targeting superenhancers in glioblastoma models. J Clin Invest. 2020;130(7):3699– 716. DOI: 10.1172/JCI129049
64. Guyon J., Fernandez-Moncada I., Larrieu C.M., Bouchez C.L., Pagano Zottola A.C., Galvis J., et al. Lactate dehydrogenases promote glioblastoma growth and invasion via a metabolic symbiosis. EMBO Mol Med. 2022;14(12):e15343. DOI: 10.15252/emmm.202115343
65. Valvona C.J., Fillmore H.L., Nunn P.B., Pilkington G.J. The Regulation and function of lactate dehydrogenase a: therapeutic potential in brain tumor. Brain Pathol. 2016;26(1):3–17. DOI: 10.1111/bpa.12299
66. Malaquin N., Carrier-Leclerc A., Dessureault M., Rodier F. DDR-mediated crosstalk between DNA-damaged cells and their microenvironment. Front Genet. 2015;6:94. DOI: 10.3389/fgene.2015.00094
67. Wang J.Y.J. Cell death response to DNA damage. Yale J Biol Med. 2019;92(4):771–9. PMID: 31866794
68. Bigge J., Koebbe L.L., Giel A.S., Bornholdt D., Buerfent B., Dasmeh P., et al. Expression quantitative trait loci influence DNA damage-induced apoptosis in cancer. BMC Genomics. 2024;25(1):1168. DOI: 10.1186/s12864-024-11068-6
69. Wu S., Li X., Gao F., de Groot J.F., Koul D., Yung W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021;23(6):920–31. DOI: 10.1093/neuonc/noab003
70. Yuan A.L., Meode M., Tan M., Maxwell L., Bering E.A., Pedersen H., et al. PARP inhibition suppresses the emergence of temozolomide resistance in a model system. J Neurooncol. 2020;148(3):463–72. DOI: 10.1007/s11060-020-03561-1
71. Xavier M.A., Rezende F., Titze-de-Almeida R., Cornelissen B. BRCAness as a biomarker of susceptibility to PARP inhibitors in glioblastoma multiforme. Biomolecules. 2021;11(8):1188. DOI: 10.3390/biom11081188
72. Meimand S.E., Pour-Rashidi A., Shahrbabak M.M., Mohammadi E., Meimand F.E., Rezaei N. The prognostication potential of BRCA genes expression in gliomas: a genetic survival analysis study. World Neurosurg. 2022;157:e123–8. DOI: 10.1016/j.wneu.2021.09.107
73. Sun P., Li Y., Chao X., Li J., Luo R., Li M., et al. Clinical characteristics and prognostic implications of BRCA-associated tumors in males: a pan-tumor survey. BMC Cancer. 2020;20(1):994. DOI: 10.1186/s12885-020-07481-1
74. Nile D.L., Rae C., Hyndman I.J., Gaze M.N., Mairs R.J. An evaluation in vitro of PARP-1 inhibitors, rucaparib and olaparib, as radiosensitisers for the treatment of neuroblastoma. BMC Cancer. 2016;16:621. DOI: 10.1186/s12885-016-2656-8
75. Parrish K.E., Cen L., Murray J., Calligaris D., Kizilbash S., Mittapalli R.K., et al. Efficacy of PARP inhibitor rucaparib in orthotopic glioblastoma xenografts is limited by ineffective drug penetration into the central nervous system. Mol Cancer Ther. 2015;14(12):2735–43. DOI: 10.1158/1535-7163.MCT-15-0553
76. van Vuurden D.G., Hulleman E., Meijer O.L., Wedekind L.E., Kool M., Witt H., et al. PARP inhibition sensitizes childhood high grade glioma, medulloblastoma and ependymoma to radiation. Oncotarget. 2011;2(12):984–96. DOI: 10.18632/oncotarget.362
77. Chornenkyy Y., Agnihotri S., Yu M., Buczkowicz P., Rakopoulos P., Golbourn B., et al. Poly-ADP-Ribose polymerase as a therapeutic target in pediatric diffuse intrinsic pontine glioma and pediatric high-grade astrocytoma. Mol Cancer Ther. 2015;14(11):2560–8. DOI: 10.1158/1535-7163.MCT-15-0282
78. Wu S., Gao F., Zheng S., Zhang C., Martinez-Ledesma E., Ezhilarasan R., et al. EGFR amplification induces increased DNA damage response and renders selective sensitivity to talazoparib (PARP Inhibitor) in glioblastoma. Clin Cancer Res. 2020;26(6):1395–407. DOI: 10.1158/1078-0432.CCR-19-2549
79. Sachdev E., Tabatabai R., Roy V., Rimel B.J., Mita M.M. PARP inhibition in cancer: an update on clinical development. Target Oncol. 2019;14(6):657–79. DOI: 10.1007/s11523-019-00680-2
80. Lin F., de Gooijer M.C., Roig E.M., Buil L.C., Christner S.M., Beumer J.H., et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res. 2014;20(10):2703–13. DOI: 10.1158/1078-0432.CCR-14-0084
81. Wagner L.M. Profile of veliparib and its potential in the treatment of solid tumors. Onco Targets Ther. 2015;8:1931–9. DOI: 10.2147/OTT.S69935
82. Barazzuol L., Jena R., Burnet N.G., Meira L.B., Jeynes J.C., Kirkby K.J., et al. Evaluation of poly (ADP-ribose) polymerase inhibitor ABT-888 combined with radiotherapy and temozolomide in glioblastoma. Radiat Oncol. 2013;8:65. DOI: 10.1186/1748-717X-8-65
83. Zhiyu Tang, Bin Jiang, Zhenyan Shi, Wenfeng Gong, Yong Liu, Xing Wang, et al. Abstract 1651: BGB-290, a novel PARP inhibitor with unique brain penetration ability, demonstrated strong synergism with temozolomide in subcutaneous and intracranial xenograft models. Cancer Res. 2015;75 (15_Suppl):1651. DOI:10.1158/1538-7445.AM2015-1651
84. Zimmermann A., Zenke F.T., Chiu L.Y., Dahmen H., Pehl U., Fuchss T., et al. A new class of selective ATM inhibitors as combination partners of DNA double-strand break inducing cancer therapies. Mol Cancer Ther. 2022;21(6):859–70. DOI: 10.1158/1535-7163.MCT-21-0934
85. Priya B., Ravi S., Kirubakaran S. Targeting ATM and ATR for cancer therapeutics: Inhibitors in clinic. Drug Discov Today. 2023;28(8):103662. DOI: 10.1016/j.drudis.2023.103662
86. Manic G., Obrist F., Sistigu A., Vitale I. Trial watch: targeting ATMCHK2 and ATR-CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015;2(4):e1012976. DOI: 10.1080/23723556.2015.1012976
87. Smith H.L., Southgate H., Tweddle D.A., Curtin N.J. DNA damage checkpoint kinases in cancer. Expert Rev Mol Med. 2020;22:e2. DOI: 10.1017/erm.2020.3
88. Blake S.M., Stricker S.H., Halavach H., Poetsch A.R., Cresswell G., Kelly G., et al. Inactivation of the ATMIN/ATM pathway protects against glioblastoma formation. Elife. 2016;5:e08711. DOI: 10.7554/eLife.08711
89. Vecchio D., Daga A., Carra E., Marubbi D., Raso A., Mascelli S., et al. Pharmacokinetics, pharmacodynamics and efficacy on pediatric tumors of the glioma radiosensitizer KU60019. Int J Cancer. 2015;136(6):1445–57. DOI: 10.1002/ijc.29121
90. Jin M.H., Oh D.Y. ATM in DNA repair in cancer. Pharmacol Ther. 2019;203:107391. DOI: 10.1016/j.pharmthera.2019.07.002
91. Chen J., Laverty D.J., Talele S., Bale A., Carlson B.L., Porath K.A., et al. Aberrant ATM signaling and homology-directed DNA repair as a vulnerability of p53-mutant GBM to AZD1390-mediated radiosensitization. Sci Transl Med. 2024;16(734):eadj5962. DOI: 10.1126/scitranslmed.adj5962
92. Lozinski M., Bowden N.A., Graves M.C., Fay M., Day B.W., Stringer B.W., et al. ATR inhibition using gartisertib enhances cell death and synergises with temozolomide and radiation in patient-derived glioblastoma cell lines. Oncotarget. 2024;15:1–18. DOI: 10.18632/oncotarget.28551
93. Peng C., Chen Z., Wang S., Wang H.W., Qiu W., Zhao L., et al. The error-prone DNA polymerase κ promotes temozolomide resistance in glioblastoma through Rad17-dependent activation of ATR-Chk1 signaling. Cancer Res. 2016;76(8):2340–53. DOI: 10.1158/0008-5472.CAN-15-1884
94. Aasland D., Götzinger L., Hauck L., Berte N., Meyer J., Effenberger M., et al. Temozolomide induces senescence and repression of DNA repair pathways in glioblastoma cells via activation of ATR-CHK1, p21, and NF-κB. Cancer Res. 2019;79(1):99–113. DOI: 10.1158/0008-5472.CAN-18-1733
95. Ganesa S., Sule A., Sundaram R.K., Bindra R.S. Mismatch repair proteins play a role in ATR activation upon temozolomide treatment in MGMT-methylated glioblastoma. Sci Rep. 2022;12(1):5827. DOI: 10.1038/s41598-022-09614-x
96. Chang K.F., Liu C.Y., Huang Y.C., Hsiao C.Y., Tsai N.M. Downregulation of VEGFR2 signaling by cedrol abrogates VEGF-driven angiogenesis and proliferation of glioblastoma cells through AKT/P70S6K and MAPK/ERK1/2 pathways. Oncol Lett. 2023;26(2):342. DOI: 10.3892/ol.2023.13928
97. Gilbert M.R., Dignam J.J., Armstrong T.S., Wefel J.S., Blumenthal D.T., Vogelbaum M.A., et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370(8):699–708. DOI: 10.1056/NEJMoa1308573
98. Carmell N., Rominiyi O., Myers K.N., McGarrity-Cottrell C., Vanderlinden A., Lad N., et al. Identification and validation of ERK5 as a DNA damage modulating drug target in glioblastoma. Cancers (Basel). 2021;13(5):944. DOI: 10.3390/cancers13050944
99. Koncar R.F., Dey B.R., Stanton A.J., Agrawal N., Wassell M.L., McCarl L.H., et al. Identification of novel RAS signaling therapeutic vulnerabilities in diffuse intrinsic pontine gliomas. Cancer Res. 2019;79(16):4026–41. DOI: 10.1158/0008-5472.CAN-18-3521
100. Tripathi S., Najem H., Mahajan A.S., Zhang P., Low J.T., Stegh A.H., et al. cGAS-STING pathway targeted therapies and their applications in the treatment of high-grade glioma. F1000Res. 2022;11:1010. DOI: 10.12688/f1000research.125163.1
101. Low J.T., Brown M.C., Reitman Z.J., Bernstock J.D., Markert J.M., Friedman G.K., et al. Understanding and therapeutically exploiting cGAS/STING signaling in glioblastoma. J Clin Invest. 2024;134(2):e163452. DOI: 10.1172/JCI163452
102. He Y., Yang Y., Huang W., Yang S., Xue X., Zhu K., et al. Manganese facilitated cGAS-STING-IFNI pathway activation induced by ionizing radiation in glioma cells. Int J Radiat Biol. 2023;99(12):1890–907. DOI: 10.1080/09553002.2023.2232011
Гареев Ильгиз Фанилевич — к.м.н., старший научный сотрудник
Республика Башкортостан, Уфа; Москва
Бейлерли Озал Арзуман оглы — к.м.н., старший научный сотрудник
Республика Башкортостан, Уфа; Москва
Румянцев Сергей Александрович — д.м.н., профессор, член-корр. РАН
Москва
Гареев И.Ф., Бейлерли О.А., Румянцев С.А. Повреждение и восстановление ДНК при глиобластоме: новые перспективы терапии. Креативная хирургия и онкология. 2025;15(2):124-138. https://doi.org/10.24060/2076-3093-2025-15-2-28-42
Gareev I.F., Beylerli O.A., Roumiantsev S.A. DNA Damage and Repair in Glioblastoma: Emerging Therapeutic Perspectives. Creative surgery and oncology. 2025;15(2):124-138. (In Russ.) https://doi.org/10.24060/2076-3093-2025-15-2-28-42
Федеральное государственное бюджетное образовательное учреждение высшего образования «Башкирский государственный медицинский университет» Министерства здравоохранения Российской Федерации
450008, Республика Башкортостан, г. Уфа, ул. Пушкина, д. 96, корп. 98
Тел./факс: +7 (347) 273-56 -97
E-mail: csurgonco@bashgmu.ru






























