



Содержание
Перейти к:
 К. В. Меньшиков,
А. В. Султанбаев,
Ш. И. Мусин,
И. Р. Рахматуллина,
И. А. Меньшикова,
Р. Р. Абдеев,
Н. И. Султанбаева,
Е. В. Попова,
Г. А. Серебренников
К. В. Меньшиков,
А. В. Султанбаев,
Ш. И. Мусин,
И. Р. Рахматуллина,
И. А. Меньшикова,
Р. Р. Абдеев,
Н. И. Султанбаева,
Е. В. Попова,
Г. А. Серебренников https://doi.org/10.24060/2076-3093-2022-12-2-139-150
Перейти к:
Рак печени остается глобальной проблемой здравоохранения, и заболеваемость этой патологией имеет тенденцию к росту во всем мире. По оценкам разных авторов к 2025 году будет выявляться ежегодно более 1 миллиона пациентов с диагнозом «рак печени». Молекулярный патогенез гепатоцеллюлярной карциномы (ГЦК) варьирует в зависимости от различных генотоксических поражений и этиологии. Хотя наши представления о патофизиологии и драйверах ГЦК имеют тенденцию к улучшению, эти понимания еще далеки для воплощения в клиническую практику. Примерно 25 % случаев ГЦК связаны с различными мутациями. Патофизиология ГЦК представляет собой сложный многоэтапный процесс. Взаимодействие различных факторов лежит в основе ранних стадий злокачественной трансформации гепатоцитов и развитии ГЦК. В целом только примерно 20–25 % пациентов с ГЦК имеют как минимум одну потенциальную драйверную мутацию. Также следует отметить, что ожирение связано с повышенным риском развития ГЦК и рака во многих органах. Несмотря на многие уже известные моменты в патогенезе развития ГЦК, еще остаются нерешенные вопросы. Современные возможности молекулярно-генетической диагностики и моделирование злокачественных опухолей на животных позволяют расширить горизонты знаний в этой области.
Меньшиков К.В., Султанбаев А.В., Мусин Ш.И., Рахматуллина И.Р., Меньшикова И.А., Абдеев Р.Р., Султанбаева Н.И., Попова Е.В., Серебренников Г.А. Гепатоцеллюлярная карцинома: этиологические факторы и механизмы развития. Обзор литературы. Креативная хирургия и онкология. 2022;12(2):139-150. https://doi.org/10.24060/2076-3093-2022-12-2-139-150
Menshikov K.V., Sultanbaev A.V., Musin Sh.I., Rakhmatullina I.R., Menshikova I.A., Abdeev R.R., Sultanbaeva N.I., Popova E.V., Serebrennikov G.A. Hepatocellular Carcinoma: Aetiology and Mechanisms of Development. A Literature Review. Creative surgery and oncology. 2022;12(2):139-150. (In Russ.) https://doi.org/10.24060/2076-3093-2022-12-2-139-150
Рак печени остается глобальной проблемой здравоохранения, и заболеваемость этой патологией имеет тенденцию к росту во всем мире [1][2]. По оценкам разных авторов к 2025 году будет выявляться ежегодно более 1 миллиона пациентов с диагнозом «рак печени» [3]. Гепатоцеллюлярная карцинома (ГЦК) — наиболее распространенная формой рака печени и составляет примерно 90 % случаев. Вирус гепатита В (ВГВ) является наиболее важным фактором риска развития ГЦК и становится причиной примерно 50 % случаев [4]. Риск развития ГЦК связывают с вирусом гепатита С (ВГС). Этот риск значительно снижается за счет достижения устойчивого вирусологического ответа на противовирусные препараты [5]. Неалкогольный стеатогепатит (НАСГ), ассоциированный с метаболическим синдромом или сахарным диабетом, становится самой быстрорастущей этиологией ГЦК, особенно на Западе [6].
Молекулярный патогенез ГЦК варьирует в зависимости от различных генотоксических поражений и этиологии. Хотя наши представления о патофизиологии и драйверах ГЦК имеют тенденцию к улучшению, эти понимания еще далеки от воплощения в клиническую практику. Примерно 25 % случаев ГЦК связаны с различными мутациями. Однако распространенность большинства мутаций составляет менее 10 %, и таким образом исследования по проверке концепции о доминирующих драйверных мутациях при ГЦК, таких как TERT, TP53 и CTNNB1, остаются не изученными и не имеющими подходов к терапии [7–9]. В настоящее время конкретные достижения в нашем понимании механизмов, лежащих в основе ГЦК, связанной с НАСГ, позволили по-новому взглянуть на вклад микроокружения опухоли, особенно иммунной системы и активации тромбоцитов, в патофизиологии этого заболевания [10][11]. Диагноз ГЦК обычно основывается на неинвазивных критериях, хотя растет потребность в молекулярной характеристике опухоли с использованием биопсии ткани в клинической практике [12][13]. Лечение ГЦК заметно улучшилось с начала 2010-х годов [8][12–14]. Резекция печени и трансплантация печени были основным методом лечения в случаях ГЦК. Тщательный отбор пациентов привел к улучшению результатов хирургического лечения и хорошему безрецидивному 10-летнему периоду после трансплантации печени [12][15]. Также применяется локальная абляция радиочастотным методом под визуальным контролем для нехирургических случаев ГЦК на ранних стадиях, несмотря на прогресс в других методиках [14]. Результаты адъювантной терапии после потенциально излечивающих подходов являются неудовлетворительными, поскольку рандомизированные клинические исследования до сих пор демонстрируют отрицательные результаты. Для промежуточной стадии ГЦК трансартериальная химиоэмболизация (ТАХЭ) — наиболее широко используемый стандарт лечения [16]. Трансартериальная радиоэмболизация (TARE) продемонстрировала эффективность в исследованиях II фазы, но не была принята в качестве основного стандарта лечения [17]. Ожидается, что другие локально-регионарные подходы к радиационной онкологии не расширят арсенал возможностей лечения в ближайшем будущем.
В настоящее время системная терапия, включая ингибиторы иммунных контрольных точек (ИКИ), ингибиторы тирозинкиназы (ИТК) и моноклональные антитела, бросили вызов использованию традиционных методов лечения ГЦК. Был отмечен значительный прогресс в развитии системной терапии за последние 5 лет, в рандомизированных исследованиях сообщается о заметном увеличении общей выживаемости и улучшении качества жизни пациентов [8]. Исторически случаи ГЦК на поздних стадиях подразумевали медиану общей выживаемости примерно 8 месяцев, а одобренная комбинация атезолизумаба (антитело кPDL1) и бевацизумаба (антитело против VEGF) увеличила этот показатель более чем в два раза [18]. Ингибиторы тирозинкиназ сорафениб и ленватиниб остаются наиболее эффективными препаратами монотерапии [19][20]. В случае прогрессирования однокомпонентные режимы терапии такими препаратами, какрегорафениб, кабозантиниб и рамуцирумаб, также показали улучшение выживаемости [21–23]. Монотерапия ингибиторами контрольных точек обеспечивает существенный клинический эффект у 15–20 % пациентов, но до настоящего времени не идентифицированы биомаркеры, характеризующие эту группу [24][25]. Более того, исследования III фазы, изучающие эффективность комбинированной терапии, то есть сочетания ИКИ с ИТК или сочетания ингибиторов PD1/PDL1 с ингибиторами CTLA4, продолжаются. Ожидается, что результаты этих испытаний полностью изменят ландшафт лечения ГЦК.
Хотелось бы остановиться на вопросах эпидемиологии ГЦК. Рак печени является шестым наиболее распространенным видом рака в мире: в 2018 году было зарегистрировано 841 080 новых случаев рака печени. ГЦК является четвертой по значимости причиной смерти от рака в глобальном масштабе [3] (рис. 1).

Рисунок 1. Заболеваемость ГЦК в зависимости от географического района и этиологии
Figure 1. HCC incidence by geographic locale and aetiology
Заболеваемость и основные этиологические факторы, участвующие в гепатоканцерогенезе, изображены на рисунке 1. Самая высокая частота ГЦК наблюдается в Восточной Азии, при этом Монголия демонстрирует самую высокую заболеваемость ГЦК в мире. Вирус гепатита В (HBV) является основным этиологическим фактором в большинстве стран Азии (кроме Японии), Южной Америки и Африки. Вирус гепатита С (ВГС) является преобладающей причиной в Западной Европе, Северной Америке и Японии, а употребление алкоголя — этиологическим фактором в Центральной и Восточной Европе. Неалкогольный стеатогепатит основной этиологии, включенный в категорию «Другое», представляет собой быстрорастущий фактор риска, который, как ожидается, станет преобладающей причиной ГЦК у пациентов с высоким уровнем доходов в ближайшем будущем [3][26][27].
Самая высокая заболеваемость и смертность от ГЦК наблюдаются в Восточной Азии и Африке, хотя имеется тенденция к увеличению этих показателей в различных регионах Европы и США [28]. Действительно, наблюдение за конечными результатами в реестре SEER демонстрирует ГЦК как самую быстрорастущую причину смерти от рака в США с начала 2000-х годов, и прогнозируется, что ГЦК станет третьей ведущей причиной смерти от рака к 2030 г., если эти тенденции сохранятся [29].
Возвращаясь к факторам риска, более 90 % случаев ГЦК возникают на фоне хронического заболевания печени. Цирроз любой этиологии является одним из важнейших факторов риска развития ГЦК [12v13]. ГЦК является основной причиной смерти у пациентов с циррозом печени с ежегодной заболеваемостью ГЦК в этой группе до 1–6 % [30]. К основным факторам риска ГЦК относятся хроническое употребление алкоголя, диабет или ожирение, НАСГ и инфицирование HBV или HCV (рис. 1). Другие меньше распространенные факторы риска ГЦК включают первичный билиарный холангит, гемохроматоз и дефицит α1-антитрипсина. Действительно, у пациентов с развивающимся циррозом печени вследствие гемохроматоза особенно высок риск ГЦК, она в течение жизни появляется у 45 % таких больных [31].
Патофизиология ГЦК представляет собой сложный, многоэтапный процесс. Взаимодействие различных факторов лежит в основе ранних стадий злокачественной трансформации гепатоцитов и развитии ГЦК. К этим факторам относятся генетическая предрасположенность, реципрокные взаимодействия между вирусными и невирусными факторами риска, клеточное микроокружение и различные иммунные клетки, а также тяжесть сопутствующих хронических заболеваний печени. Измененное микроокружение является ключевой характеристикой рака. Известно, что микроокружение участвует во всех стадиях злокачественного роста, от начальных фаз трансформации до инвазивного рака, и в конечном счете приводит к метастазированию. В нашем обзоре проводится попытка детализировать текущее понимание механизмов, лежащих в основе ГЦК связанной с НАСГ. Одним из компонентов понимания развития ГЦК является так называемая ячейка происхождения. Аналогично любому типу рака исходная клетка может быть стволовой клеткой печени, транзитной амплифицирующей популяцией или зрелым гепатоцитом. В целом наличие и роль стволовых клеток в печени само по себе спорно. Кроме того, зрелые гепатоциты — это долгоживущие клетки, и они сохраняют значительную пролиферативную возможность в ответ на травму. Многие мышиные модели подтверждают возможность того, что ГЦК возникает в трансформированных клетках зрелых гепатоцитов, хотя некоторые авторы полагают, что стволовые клетки печени могут быть источником развития ГЦК [32]. Как это ни парадоксально, внутрипеченочные холангиокарциномы и опухоли со смешанной морфологией ГЦК часто возникают из зрелых гепатоцитов, что подчеркивает концепции метаплазии и клеточной пластичности (что и есть трансдифференцировка). Этот вывод подтверждает представление о том, что морфология и эпигенетический ландшафт опухоли не обязательно отражает исходную клетку [33][34].
Рассмотрим мутации генов — драйверов рака при ГЦК. Использование метода высокопроизводительного секвенирования нового поколения позволило идентифицировать гены — драйверы рака с онкогенными функциями или функциями подавления опухолей, которые периодически встречаются при ГЦК. Активация теломеразы через мутации промотора TERT, вирусные вставки, хромосомная транслокация или амплификация генов — наиболее частые изменения генов-драйверов, наблюдаемые у примерно 80 % случаев ГЦК [7][35]. Исследования продемонстрировали, что активация сигнального пути Wnt-β-катенин в30–50 % случаев вызвана мутациями в CTNNB1 (кодирует β-катенин), AXIN1 или APC (ингибиторы Wnt пути) инактивации [7][35]. Другие частые мутации или генетические изменения обнаружены в TP53, RB1, CCNA2, CCNE1, PTEN, ARID1A, ARID2, RPS6KA3 или NFE2L2, все они изменяют регуляцию клеточного цикла. Кроме того, варианты в генах, участвующих в эпигенетической регуляции, окислительном стрессе и путях AKT-mTOR и MAPK, были вовлечены в развитие ГЦК. И повторяющиеся фокальные амплификации хромосом в CCND1, FGF19, VEGFA, MYC или MET, приводящие к сверхэкспрессии, вызывают активацию различных онкогенных сигнальных путей, в том числе рецепторные тирозинкиназы [36]. Хотя мутации в гене — драйвере рака накапливаются случайным образом, определенные гены связаны с точными молекулярными подклассами ГЦК, определяемыми транскриптомными профилями и гистологическими фенотипами [8][9][37] (рис. 2).
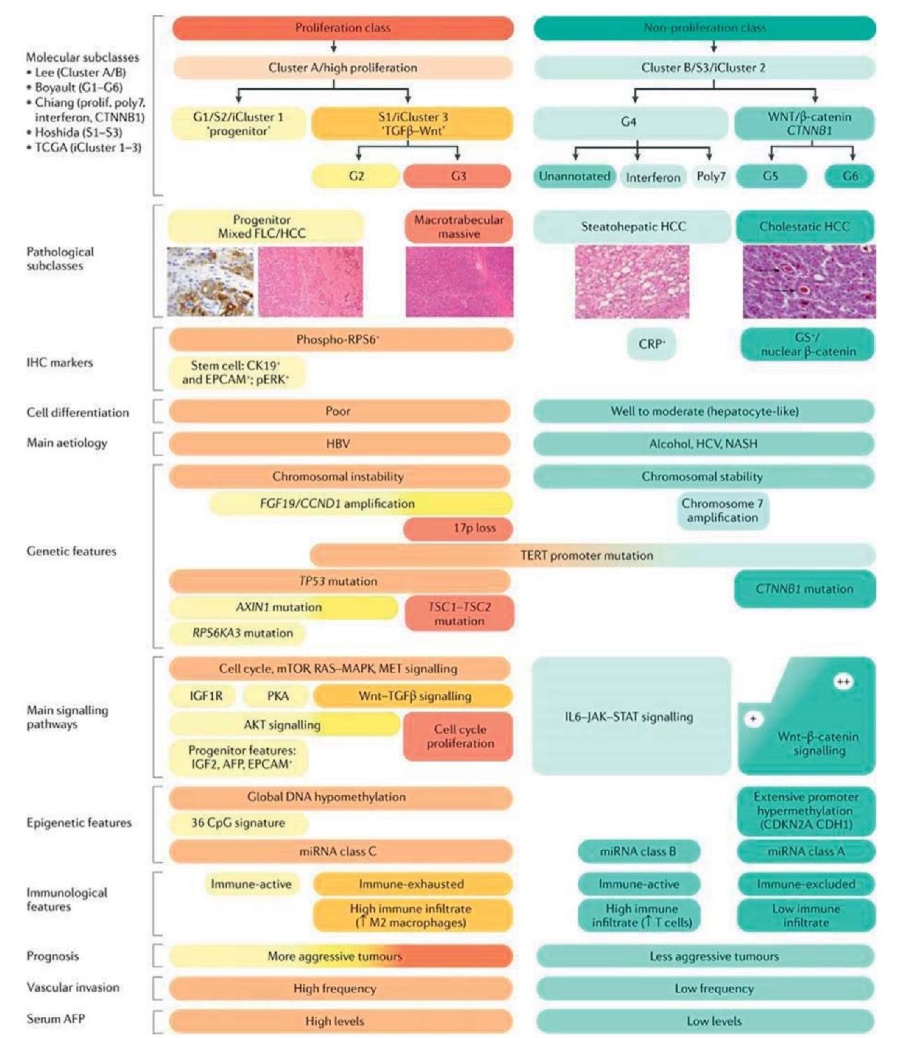
Рисунок 2. Молекулярно-иммунная классификация ГЦК
Figure 2 . Molecular and immune classification of HCC
Все варианты ГЦК можно разделить на две основные молекулярные группы на основе транскриптомных фенотипических классов [36][39–41]. Класс пролиферации характеризуется более агрессивной опухолью с низкой гистологической дифференцировкой, высокой сосудистой инвазией и повышенным уровнем α-фетопротеина (АФП) [37]. Этот класс можно разделить на два подкласса: S1, или iCluster 3, характеризующийся активацией Wnt-TGFβ, которая вызывает истощение иммунного фенотипа; и S2, или iCluster 1, характеризующийся предшественникоподобным фенотипом, с экспрессией маркеров стволовых клеток (CK19, EPCAM) и активированным IGF2 и сигнальным путем EPCAM53 [38][39][42]. Наличие опухолей, ассоциированных с вирусом гепатита В (HBV), — частая активация классических путей пролиферации клеток, таких как PI3K-AKT-mTOR, каскадов PI3K-AKT-mTOR, RAS-MAPK, MET и IGF. Кроме того, частые мутации TP53, высокая хромосомная нестабильность и глобальное гипометилирование ДНК представляют собой дополнительные признаки этого класса. Класс нераспространения характеризуется менее агрессивными опухолями с гистологической дифференциацией от хорошей до умеренной, низким уровнем АФПи менее частой сосудистой инвазией. Эти опухоли вызваны неалкогольным стеатогепатитам (НАСГ), алкогольным стеатогепатитом и инфекцией, вызванной вирусом гепатита С (ВГС). В этом классе были охарактеризованы отдельные подгруппы: WNT-β-катенин — подкласс CTNNB1 характеризуется частыми мутациями CTNNB1 и активацией WNT-сигнального пути β-катенина, который приводит к иммуноисключенному фенотипу с низкой иммунной инфильтрацией [36][41][42], а подкласс интерферона представляет собой высокоактивированный сигнальный путь IL6-JAK-STAT с более воспаленным микроокружением опухоли. Этот класс представляет хромосомную стабильность с частыми мутациями промотора TERT [1][7–9][38–42].
В целом только примерно 20–25 % пациентов с ГЦК имеют как минимум одну потенциальную драйверную мутацию в соответствии с текущими стандартами. Мутации, вызывающие злокачественный рост, взаимодействие факторов риска, хорошо описаны в патогенезе ГЦК. Например, токсическое действие афлатоксина B1 потенцируется инфекцией HBV, особенно у пациентов с нулевым полиморфизмом GSTT1 [43][44]. Кроме того, было установлено, что полиморфизмы в PNPLA3, TM6SF2 и HSD17B13 связаны с тяжестью течения НАСГ и заболеваемостью ГЦК, особенно у пациентов с высоким хроническим потреблением алкоголя [45][46].
Наиболее частая локализация HBV-опосредованного инсерционного мутагенеза находится внутри промотора TERT, который приводит к гиперэкспрессии теломеразы, фермента, отвечающего за поддержание длины теломер [47]. Активация теломеразы предотвращает эрозию хромосом, которая физиологически возникает при каждом клеточном делении при старении. Эктопическая активация теломеразы защищает клетки от старения и способствует трансформации [48]. Другие повторяющиеся вставки, связанные с HBV, были идентифицированы для активации мощных онкогенов, таких как CCNA2 или CCNE1, участвующих в контроле клеточного цикла. Эти онкогенные изменения вызывают репликативный стресс и сложные перестройки по всему геному [49]. В небольшой группе пациентов с ГЦК, аденоассоциированным вирусом 2, продемонстрирован аналогичный инсерционный онкогенный мутагенез с общей горячей точкой вирусной инсерции внутри промотора TERT, CCNA2 и CCNE1 [50]. Эти наблюдения показывают, что специфические онкогены, активированные вирусной инфекцией, действуют как ранние помощники трансформации гепатоцитов. Напротив, инфекция ВГС не может привести к сильному, прямому онкогенному эффекту и индукции мутаций в результате окислительного стресса, вызванного хроническим воспалением.
При развитии хронических заболеваний печени и цирроза, которые лежат в основе возникновения ГЦК, в большинстве случаев находятся гепатоциты, постепенно накапливаются многочисленные генетические мутации и эпигенетические изменения. По ходу этого процесса несколько факторов риска, вызывающих мутации ДНК, связаны со специфическими мутационными сигнатурами [7][51].
Несколько исследований, основанных на геномном, эпигеномном, гистопатологическом и иммунологическом анализе, установили молекулярную и иммунную классификацию ГЦК [1][9][52]. Данные представлены на рисунке 2. Определены молекулярные классы ГЦК на основе главных молекулярных драйверов и вовлеченных сигнальных путей [9][38–41] или в зависимости от иммунного статуса опухоли [8][42]. Эти молекулярные классы коррелируют со специфическими геномными нарушениями, гистопатологическими особенностями и клиническими исходами. Для примерно 50 % случаев ГЦК и в целом имеются мутации в гене TP53 и в амплификациях FGF19 или CCND1 [36]. Кроме того, данные мутации чаще встречаются при ВГВ-ассоциированном ГЦК и имеют худший прогноз. Данный класс включает два подкласса: группу клеток — предшественников пролиферации и группу клеток — предшественников пролиферации Wnt-TGFβ. Группа клеток — предшественников пролиферации, которая составляет 25–30 %, характеризуется ГЦК c активацией классических путей пролиферации клеток (таких как как сигнальный путь PI3K-AKT-mTOR, путь RAS-MAPK и сигнальные каскады MET и IGF9 ) и экспрессией маркеров клеток-предшественников (таких, как EPCAM и α-фетопротеин) и соответствует кластеру 1 [36][38]. Группа WNT-TGFβ, на которую приходится 20 % случаев ГЦК, характеризуется неканонической активацией Wnt и коррелирует с кластером 3 TCGA. И наоборот, непролиферативный класс опухоли, на долю которого приходится 50 % ГЦК, чаще встречается при ГЦК, связанной с употреблением алкоголя, и ГЦК, связанной с ВГС; эти опухоли коррелируют с кластером 2 TCGA [39].
В пределах непролиферативного класса по крайней мере две отдельные подгруппы были определены: одна характеризуется доминантной канонической передачей сигналов Wnt, связанной с мутациями в CTNNB1 (ref.72), а вторая характеризуется путем активации передачи сигналов IFNα [36][53]. Отчеты о классификации ГЦК согласно статусу иммунных клеток еще больше расширили наше понимание молекулярных признаков ГЦК (рис. 2) [42]. Эта классификация предоставляет дополнительную информацию, основанную на иммунных признаках, и делит варианты ГЦК на отчетливые подклассы: иммуноактивные, иммуноистощенные, иммунно-промежуточные и иммунно-исключенные. Иммунный класс, который включает как иммуноактивные, так и иммуноистощенные подклассы, характеризуется инфильтратами иммунных клеток различной природы. Иммуноактивные опухоли ГЦК (выявляются в 20 % случаев) обогащены активными Т-хелперами (CD4+), клеточными инфильтратами и цитотоксическими Т (CD8+) клетками, инфильтрирующими и реагирующими к ингибиторам контрольных точек. И наоборот, опухоли, где преобладает состояние истощения клеток CD8+, вызванное TGF-β, иммуноистощенные опухоли, которые представляют собой другой конец спектра, характеризуются недостатком инфильтрирующих Т-клеток и увеличением регуляторных Т-(Treg) клеток, и в них доминируют канонические сигналы Wnt и другие иммунно-дрессуативные каскады. Иммуноисключенные опухоли в первую очередь устойчивы к ингибиторам контрольных точек [54].
Ожирение связано с повышенным риском развития рака во многих органах [55]. Ожирение может вызвать системные изменения, включая измененную иммунную функцию и системные эндокринные изменения, которые являются отличительными чертами нескольких типов злокачественных новообразований. Имеющиеся данные показывают, что жировая болезнь печени быстро становится ведущей причиной ГЦК на Западе [6]. Исследования показали, что специфические для печени механизмы, посредством которых неалкогольная жировая болезнь печени или НАСГ способствуют развитию ГЦК, включают метаболический и окислительный стресс, измененную иммунную функцию, патологические воспалительные реакции и измененную эндокринную передачу сигналов [10][56].
Гепатоциты, перегруженные жирными кислотами, вызывают окислительный стресс и эндоплазматический ретикулярный (ЭР) стресс, вызывающий патологическое воспаление и повреждение клеток [10][11]. Одно исследование доказало причинную роль ЭР стресса при НАСГ-индуцированном ГЦР у мышей; ЭР стресс в гепатоцитах мышей приводил к активации воспалительных сигнальных путей, в частности NF-kB и TNF, приводя к индукции ГЦК [57]. Однако эти патогенные механизмы еще предстоит доказать в роли развития ГЦК у человека. Нарушение метаболизма жирных кислот в гепатоцитах может вызывать повреждение ДНК из-за увеличения количества активных форм кислорода (АФК), образующихся в результате дисфункции митохондрий [58]. При НАСГ образование липидов может не только увеличиваться, но, возможно, изменяться, чтобы генерировать больше патогенных липидов, которые служат онкометаболитами [59][60]. Например, постоянная активация mTORC2 в гепатоцитах мышей увеличивала образование сфинголипида глюкозилцерамида, вызывая повышенную продукцию АФК, что в конечном итоге может привести к развитию ГЦК [59]. Точно так же измененный метаболизм холестерина также может способствовать патогенезу ГЦК, потенциально за счет продукции проонкогенных лигандов ядерных рецепторов [60].
Хотя аутофагия может иметь противоопухолевые функции, одно исследование продемонстрировало важную роль липофагии (то есть аутофагической деградации липидных капель) в патогенезе ГЦК. Сверхэкспрессия секвестосомы 1 (также известной как p62), которая регулирует липофагию в гепатоцитах больных сНАСГ и в мышиной модели, была связана с развитием ГЦК [61]. Исследования показали более высокий риск ГЦК у пациентов с НАСГ, чем у пациентов с неалкогольной жировой болезнью печени [6]. Одно экспериментальное исследование показало, что окислительные реакции, вызванные жирными кислотами, обуславливаю стресс в гепатоцитах, в свою очередь активируют STAT1 и STAT3, оба они являются провоспалительными транскрипционными факторами, которые обычно действуют параллельно [62]. Примечательно, что в этой мышиной модели высокие уровни STAT1 вызывали прогрессирование НАСГ, в то время как высокие уровни STAT3 способствовали развитию ГЦК, независимо друг от друга [62]. Это говорит о том, что сходные воспалительные сигналы могут по-разному способствовать прогрессированию неалкогольной болезни печени в НАСГ или ГЦК. Поскольку неалкогольная болезнь печени более распространена, чем НАСГ, в общей популяции, это открытие подчеркивает необходимость лучшего понимания того, как неалкогольная болезнь печени сама по себе, независимо от НАСГ, может перейти в ГЦК [6]. В совокупности ЭР стресс, патологическая липофагия, увеличение производства АФК и уменьшение мощности восстановления (низкий уровень NADH или NADPH) могут вызвать онкогенные генетические изменения в перегруженных жирными кислотами гепатоцитах и способствуют размножению злокачественных клеток.
Инфильтрация иммунными клетками жировой ткани печени является гистопатологическим признаком НАСГ [10]. Разработка моделей животных, точно воспроизводящих ГЦР человека, необходима для фундаментальных исследований изучения патогенеза и трансляционных исследований [63–77]. Данные представлены на рисунке 3.

Рисунок 3. Доклинические и клинические модели, используемые в трансляционных исследованиях ГЦК
Figure 3. Preclinical and clinical models in translational HCC studies
Трансляционные исследования в ГЦК представляют собой дорогу с двусторонним движением между доклиническими и клиническими моделями. С одной стороны, доклинические модели помогают понять патогенез и механизмы, участвующие в инициировании и прогрессировании заболевания, и формируют основу для разработки клинических методов лечения. Например, клеточные линии обеспечивают быструю, относительно простую, но менее актуальную клиническую информацию, в то время как модели ксенотрансплантата пациента медленны, сложны, но более актуальны. Клинические исследования сосредоточены на разработке лекарств и открытии биомаркеров, а их результаты, хотя и отрицательные, часто приводят к новым гипотезам, требующим доклинического исследования. Исследования I фазы направлены на понимание фармакокинетики и профилей токсичности недавно разработанных лекарств, исследования II фазы предназначены для изучения предварительной эффективности, а исследования III фазы, рандомизированные контролируемые испытания, представляют самый высокий уровень доказательств, необходимых для одобрения регулирующими органами. Биомаркеры позволяют проводить отбор популяций, которые с наибольшей вероятностью выиграют от определенных видов лечения на основе их механизма действия.
Несколько экспериментальных моделей показали, что иммунные клетки и цитокины играют важную роль в патогенезе ГЦК. Например, длительный НАСГ у мышиных моделей индуцирует активацию CD8+ Т-клеток, что приводит к повреждению гепатоцитов и развитию ГЦК [78]. Кроме того, неалкогольная жировая болезнь печени вызывает избирательную потерю внутрипеченочных CD4+ клеток, которые имеют решающее значение для индукции эффективного противоопухолевого адаптивного иммунного ответа [79]. Другие иммунные типы клеток, включая В-клетки, Treg-клетки, естественные клетки-киллеры и различные типы миелоидных клеток, связаны с патогенезом ГЦК, индуцированным НАСГ [10][56]. Интересно, что в соответствии с клиническими данными рекрутирование и активация тромбоцитов в печени также способствуют развитию ГЦК у мышей, особенно путем передачи сигналов гликопротеина тромбоцитов Ib-α (GPIb-α), что предполагает терапевтический потенциал этого пути [80]. Было также показано, что цитокиновая среда лежит в основе причинной роли НАСГ в ГЦК [11]. Все вышеописанные механизмы могут одновременно способствовать развитию ГЦК на фоне жировой дистрофии печени. Однако их относительный вклад в человеческую ГЦК в настоящее время неизвестен. Анализ мутационных подтипов в ГЦК, связанной с НАСГ, по сравнению с ГЦК других этиологий, мог бы помочь определить относительный вклад различных факторов.
ГЦК является злокачественной опухолью, связанной с воспалением, при этом ~ 90 % случаев ГЦК связано с длительным воспалением вследствие вирусного гепатита, чрезмерного употребления алкоголя, неалкогольной жировой болезни печени или НАСГ. Иммунное микроокружение играет ключевую роль в патогенезе ГЦК [81]. При ГЦК наличие иммунных инфильтратов связано с лучшим прогнозом, вероятно, благодаря более эффективному противоопухолевому иммунитету [58][82]. Мышиные модели ГЦК показали, что иммунные сигналы, такие как IL-6, лимфотоксин-α и TNF, могут ускорять гепатоканцерогенез и воздействуют на агрессивность опухоли [34][83]. Тем не менее иммунный ответ также ограничивает прогрессирование рака печени [81]. Важно отметить, что печень содержит наибольшее количество иммунных клеток в организме и поддерживает уникальный иммунный статус, значительно более толерантный, чем другие органы, что позволяет ему противостоять постоянному потоку воспалительных сигналов из кишечника [81]. Понимание этой уникальной печеночной иммунной системы, вероятно, важно в контексте сложного взаимодействия между злокачественными гепатоцитами и иммунной системой печени [81][84]. Примечательно, что исследования на мышах и людях позволяют предположить, что фактор VEGF, секретируемый злокачественными гепатоцитами, образует иммунотолерантную проонкогенную микросреду, предполагая, что блокирование каскада VEGF-может быть эффективным путем изменения иммунной толерантности печени [36][85]. Интересно, что комбинации ингибиторов контрольных точек со специфическими таргетными препаратами, такими как VEGF-ингибиторы, показали более хорошие результаты в лечении ГЦК, чем использование отдельных препаратов [19][86]. В хронически воспаленной печени многие клеточные типы, в том числе макрофаги, звездчатые клетки, эндотелиальные клетки и различные подтипы лимфоцитов, взаимодействуют с гепатоцитами [81][84]. Пониманию роли адаптивной иммунной системы уделяется повышенное внимание в связи с ее важностью в иммуноонкологии. В частности, выводы на основании исследований на моделях мышей показывают, что практически каждый тип иммунных клеток может иметь как проопухолевую, так и противоопухолевую роль [81] — два основных проонкогенных механизма, посредством которых иммунные клетки способствуют развитию ГЦК, включая секрецию цитокинов и факторы роста. Эти механизмы способствуют пролиферации или противодействуют апоптозу опухолевых клеток, а также, как это ни парадоксально, подавлению противоопухолевой функции соседних лимфоцитов. Исследования показали, что пути NF-κB и JAK-STAT являются ключевыми воспалительными сигнальными путями, участвующими в промотировании ГЦК [87]. Этот вывод получил дальнейшее подтверждение в анализе транскриптома ГЦК человека [84]. Главная противоопухолевая функция адаптивной иммунной системы опосредуется через иммунный надзор и элиминацию предраковыхили полностью трансформированных злокачественных гепатоцитов [82].
Цитотоксические Т (CD8+) клетки считаются ключевыми эффекторами противоопухолевого иммунитета. Соответственно, одно исследование показало, что их истощение у мышей может увеличить риск развития ГЦК, и другое исследование показало, что эти Т-клетки опосредуют надзор за предраковыми гепатоцитами [89]. Как это ни парадоксально, в нескольких конкретных случаях истощение CD8+ Т-клеток у мышей привело к уменьшению опухолевой нагрузки, что указывает на то, что эти клетки также могут иметь проонкогенные функции. Анализы образцов ГЦК человека выявили наличие функциональных CD8+Т-клеток, экспрессирующих противоопухолевые эффекторные молекулы, такие как гранзим А, гранзим В и перфорин, у некоторых пациентов [91]. Тем не менее одноклеточное РНК секвенирование Т-клеток в ГЦК человека предполагает, что во многих случаях эти CD8+ Т-клетки дисфункциональны [92]. Причины дисфункции CD8+ Т-клеток, проявляющиеся снижением пролиферации и уменьшенной способностью продуцировать цитотоксические эффекторные молекулы, недостаточно выяснены.
Treg-клетки считаются главными виновниками дисфункции Т-клеток при ГЦК, и наличие их в большом количестве в опухоли связано с более неблагоприятными исходами заболевания [93]. Иммунодепрессивные функции Treg-клеток могут быть опосредованы секрецией CD10 и TGF-β116, если предполагать, что нацеливание на эти цитокины может повышать чувствительность ГЦК к ингибиторам контрольных точек. Интересно, что рецептор гиалуроновой кислоты, лайилин, был связан с супрессивной функцией Treg-клеток, инфильтрирующих ГЦК. Индукция лайилина вызывает дисфункцию CD8+ Т-клеток при ГЦК человека, и его гиперэкспрессия в лимфоцитах человека связана с уникальной сигнатурой экспрессии мРНК [92].
Хотя В-клетки считались ранее не принимающими участие в развитии рака, новые данные подтверждают их активное участие в перекрестных помехах между адаптивной иммунной системой и раком [94]. В мышиной модели ГЦК В-клетки как активируют, так и подавляют рост опухоли [95]. Кроме того, одно исследование показало, что IgA-экспрессирующие лимфоциты поддерживают рост ГЦК за счет активного подавления функции CD8+ Т-клеток [92]. Наконец, исследования на людях и мышах показали, что третичные лимфоидные структуры, играющие важную роль в адаптивном иммунном ответе на злокачественную опухоль, продемонстрировали способность к проопухолевому и противоопухолевому ответу при ГЦК [96–98]. Таким образом, третичные лимфоидные структуры, подобные макрофагам и лимфоцитам, могут являться либо противоопухолевыми, либо проонкогенными компонентами при ГЦК.
Несмотря на то что некоторые этиологии с большей вероятностью вызывают ГЦК, чем другие (например, ВГС против аутоиммунного гепатита), как только пациент достигает цирротической стадии, риск ГЦК достаточен для возмещения затрат на эффективный скрининг [12][13]. Ключевой клеткой, участвующей в реакции печени на хроническое повреждение, является звездчатая клетка печени, которая при активации претерпевает фенотипические изменения и синтезирует компоненты внеклеточного матрикса, в основном коллагена и факторов роста, которые способствуют миграции эндотелиальных клеток, неоангиогенезу и фиброзу [99][100]. Последующее искажение печеночной архитектуры и дезорганизованная сосудистой сети являются гистологическим субстратом цирроза и портальной гипертензии. В ответ предраковые стареющие гепатоциты секретируют хемокины, препятствующие надзору за старением, и нарушают иммуноопосредованное подавление опухоли в естественных условиях [90]. Кроме того, экспериментальные модели подтвердили важность CD4+ лимфоцитов в развитии ГЦК [79], связанной с неалкогольной болезнью печени, а также взаимодействие между врожденной иммунной системой и кишечной микрофлорой, что способствует развитию ГЦК [101][102]. Таким образом, помимо фиброза, иммунная система вносит существенный вклад в эффект поля рака при ГЦК. Пермиссивную микросреду при циррозе, способствующую развитию опухоли, обычно называют эффектом поля рака. Различные геномные исследования охарактеризовали доминирующие молекулярные элементы, разрегулированные в этом микроокружении. Многочисленные сигнатуры генов, полученные из цирротической ткани, коррелируют с риском развития ГЦК и могут быть использованы для стратификации риска пациентов [88][103][104]. Эти сигнатуры генов коррелируют с риском развития рака, а также с вероятностью заболевания печени в стадии декомпенсации и общей выживаемостью [103][104]. В большом количестве исследований подробно описаны геномные признаки воспалительного микроокружения при циррозе печени, способствующие развитию ГЦК [105]. Подкласс иммунодепрессантов, который продемонстрировал усиление в передаче сигналов TGF-β, истощение Т-клеток и гиперэкспрессию иммунных контрольных точек (таких как CTLA4, TIGIT, LAG3), был выявлен примерно у 10 % пациентов, которые имели более высокий риск развития ГЦК (увеличение риска в 3 раза через 5 и 10 лет) [105]. Решающую роль играет микроокружение опухоли. Окружающая среда в естественном течении ГЦК является убедительным обоснованием для модуляции динамических перекрестных помех между гепатоцитами и иммунной системой печени в качестве терапевтической стратегии [81].
Рак печени остается глобальной проблемой здравоохранения, и заболеваемость этой патологией имеет тенденцию к росту во всем мире. Несмотря на многие уже известные моменты в патогенезе развития ГЦК, еще остаются нерешенные вопросы. Современные возможности молекулярно-генетической диагностики и моделирование злокачественных опухолей на животных позволяют расширить горизонты знаний в этой области. В приведенном обзоре литературы мы постарались продемонстрировать основные открытия в области этиологии и патогенезе развития ГЦК. Безусловно, знания о патогенезе ГЦК позволят внедрить уже в ближайшем будущем новые терапевтические опции для лечения этой патологии. Очевидно, что продолжение исследований в этой области может переломить тренд неблагоприятного течения ГЦК в современном мире.
Информация о конфликте интересов. Конфликт интересов отсутствует.
Conflict of Interest . The authors declare no conflict of interest.
Информация о спонсорстве. Данная работа не финансировалась.
Sponsorship Data . This work is not funded.
1. Llovet J.M., Kelley R.K., Villanueva A., Singal A.G., Pikarsky E., Roayaie S., et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. DOI: 10.1038/s41572-020-00240-3
2. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–62. DOI: 10.1056/NEJMra1713263
3. Cancer Online. International Agency for Research on Cancer. GLOBOCAN 2018. Available at: https://gco.iarc.fr/today/online-analysismap?v=2020&mode=population&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=11&type=0&statistic=5&prevalence=0&population_groupearth&color_palette=default&map_scale=quantile&map_nb_colors=5&continent=0&rotate= %255B10 %252C0 %255D (accessed 20 July 2020).
4. Akinyemiju T., Abera S., Ahmed M., Alam N., Alemayohu M.A., Allen C., et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 2015. JAMA Oncol. 2017;3(12):1683–91. DOI: 10.1001/jamaoncol.2017.3055
5. Kanwal F., Kramer J., Asch S.M., Chayanupatkul M., Cao Y., El-Serag H.B. Risk of hepatocellular cancer in HCV patients treated with directacting antiviral agents. Gastroenterology. 2017;153(4):996–1005.e1. DOI: 10.1053/j.gastro.2017.06.012
6. Estes C., Razavi H., Loomba R., Younossi Z., Sanyal A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67(1):123–33. DOI: 10.1002/hep.29466
7. Schulze K., Imbeaud S., Letouzé E., Alexandrov L.B., Calderaro J., Rebouissou S., et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–11. DOI: 10.1038/ng.3252
8. Llovet J.M., Montal R., Sia D., Finn R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599–616.
9. Zucman-Rossi J., Villanueva A., Nault J.C., Llovet J.M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015;149(5):1226–39.e4. DOI: 10.1053/j.gastro.2015.05.061
10. Anstee Q.M., Reeves H.L., Kotsiliti E., Govaere O., Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16(7):411–28. DOI: 10.1038/s41575-019-0145-7
11. Friedman S.L., Neuschwander-Tetri B.A., Rinella M., Sanyal A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–22. DOI: 10.1038/s41591-018-0104-9
12. European Association for the Study of the Liver. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236. DOI: 10.1016/j.jhep.2018.03.019
13. Marrero J.A., Kulik L.M., Sirlin C.B., Zhu A.X., Finn R.S., Abecassis M.M., et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the study of liver diseases. Hepatology. 2018;68(2):723–50. DOI: 10.1002/hep.29913
14. Llovet J.M., De Baere T., Kulik L., Haber P.K., Greten T.F., Meyer T., et al. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18(5):293–313. DOI: 10.1038/s41575-020-00395-0
15. Tabrizian P., Holzner M., Halazun K., Agopian V.G., Busuttil R.W., Yao F., et al. A US multicenter analysis of 2529 HCC patients undergoing liver transplantation: 10-year outcome assessing the role of down-staging to within Milan criteria [abstract 15]. Hepatology. 2019;70:10–1.
16. Llovet J.M., Bruix J. Systematic review of randomized trials for unresectable hepatocellular carcinoma: Chemoembolization improves survival. Hepatology. 2003;37(2):429–42. DOI: 10.1053/jhep.2003.50047
17. Salem R., Gordon A.C., Mouli S., Hickey R., Kallini J., Gabr A., et al. Y90 Radioembolization significantly prolongs time to progression compared with chemoembolization in patients with hepatocellular carcinoma. Gastroenterology. 2016;151(6):1155–63.e2. DOI: 10.1053/j.gastro.2016.08.029
18. Finn R.S., Qin S., Ikeda M., Galle P.R., Ducreux M., Kim T.Y., et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–905. DOI: 10.1056/NEJMoa1915745
19. Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F., et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90. DOI: 10.1056/NEJMoa0708857
20. Kudo M., Finn R.S., Qin S., Han K.H., Ikeda K., Piscaglia F., et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 noninferiority trial. Lancet. 2018;391(10126):1163–73. DOI: 10.1016/S0140-6736(18)30207-1
21. Bruix J., Qin S., Merle P., Granito A., Huang Y.H., Bodoky G., et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. DOI: 10.1016/S0140-6736(16)32453-9
22. Shlomai A., Leshno M., Goldstein D.A. Cabozantinib for patients with advanced hepatocellular carcinoma: a cost-effectiveness analysis. Therap Adv Gastroenterol. 2019;12:1756284819878304. DOI: 10.1177/1756284819878304
23. Zhu A.X., Kang Y.K., Yen C.J., Finn R.S., Galle P.R., Llovet J.M., et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(2):282–96. DOI: 10.1016/S1470-2045(18)30937-9
24. El-Khoueiry A.B., Sangro B., Yau T., Crocenzi T.S., Kudo M., Hsu C., et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502. DOI: 10.1016/S0140-6736(17)31046-2
25. Finn R.S., Ryoo B.Y., Merle P., Kudo M., Bouattour M., Lim H.Y., et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, doubleblind, phase iii trial. J Clin Oncol. 2020;38(3):193–202. DOI: 10.1200/JCO.19.01307
26. Singal A.G., Lampertico P., Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J Hepatol. 2020;72(2):250–61. DOI: 10.1016/j.jhep.2019.08.025
27. El Dika I., Makki I., Abou-Alfa G.K. Hepatocellular carcinoma, novel therapies on the horizon. Chin Clin Oncol. 2021;10(1):12. DOI: 10.21037/cco-20-113.
28. McGlynn K.A., Petrick J.L., London W.T. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis. 2015;19(2):223–38. DOI: 10.1016/j.cld.2015.01.001
29. Rahib L., Smith B.D., Aizenberg R., Rosenzweig A.B., Fleshman J.M., Matrisian L.M. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21. DOI: 10.1158/0008-5472.CAN-14-0155
30. Trinchet J.C., Bourcier V., Chaffaut C., Ait Ahmed M., Allam S., Marcellin P., et al. Complications and competing risks of death in compensated viral cirrhosis (ANRS CO12 CirVir prospective cohort). Hepatology. 2015;62(3):737–50. DOI: 10.1002/hep.27743
31. Fracanzani A.L., Conte D., Fraquelli M., Taioli E., Mattioli M., Losco A., et al. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology. 2001;33(3):647–51. DOI: 10.1053/jhep.2001.22506
32. Sia D., Villanueva A., Friedman S.L., Llovet J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017;152(4):745–61. DOI: 10.1053/j.gastro.2016.11.048
33. Pikarsky E. Neighbourhood deaths cause a switch in cancer subtype. Nature. 2018;562(7725):45–6. DOI: 10.1038/d41586-018-06217-3
34. Seehawer M., Heinzmann F., D’Artista L., Harbig J., Roux P.-F., Hoenicke L., et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562:69–75.
35. Guichard C., Amaddeo G., Imbeaud S., Ladeiro Y., Pelletier L., Maad I.B., et al. Integrated analysis of somatic mutations and focal copynumber changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–8. DOI: 10.1038/ng.2256
36. Chiang D.Y., Villanueva A., Hoshida Y., Peix J., Newell P., Minguez B., et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68(16):6779–88. DOI: 10.1158/0008-5472.CAN-08-0742
37. Calderaro J., Ziol M., Paradis V., Zucman-Rossi J. Molecular and histological correlations in liver cancer. J Hepatol. 2019;71(3):616–30. DOI: 10.1016/j.jhep.2019.06.001
38. Hoshida Y., Nijman S.M., Kobayashi M., Chan J.A., Brunet J.P., Chiang D.Y., et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69(18):7385–92. DOI: 10.1158/0008-5472.CAN-09-1089
39. Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327-1341.e23. DOI: 10.1016/j.cell.2017.05.046
40. Lee J.S., Heo J., Libbrecht L., Chu I.S., Kaposi-Novak P., Calvisi D.F., et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12(4):410–6. DOI: 10.1038/nm1377
41. Boyault S., Rickman D.S., de Reyniès A., Balabaud C., Rebouissou S., Jeannot E., et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45(1):42–52. DOI: 10.1002/hep.21467
42. Sia D., Jiao Y., Martinez-Quetglas I., Kuchuk O., Villacorta-Martin C., Castro de Moura M., et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology. 2017;153(3):812–26. DOI: 10.1053/j.gastro.2017.06.007
43. Bressac B., Kew M., Wands J., Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350(6317):429–31. DOI: 10.1038/350429a0
44. Wang B., Huang G., Wang D., Li A., Xu Z., Dong R., et al. Null genotypes of GSTM1 and GSTT1 contribute to hepatocellular carcinoma risk: evidence from an updated meta-analysis. J Hepatol. 2010;53(3):508–18. DOI: 10.1016/j.jhep.2010.03.026
45. Romeo S., Kozlitina J., Xing C., Pertsemlidis A., Cox D., Pennacchio L.A., et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–5. DOI: 10.1038/ng.257
46. Buch S., Stickel F., Trépo E., Way M., Herrmann A., Nischalke H.D., et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47(12):1443–8. DOI: 10.1038/ng.3417
47. Paterlini-Bréchot P., Saigo K., Murakami Y., Chami M., Gozuacik D., Mugnier C., et al. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22(25):3911–6. DOI: 10.1038/sj.onc.1206492
48. Nault J.C., Ningarhari M., Rebouissou S., Zucman-Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol. 2019;16(9):544–58. DOI: 10.1038/s41575-019-0165-3
49. Bayard Q., Meunier L., Peneau C., Renault V., Shinde J., Nault J.C., et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat Commun. 2018;9(1):5235. DOI: 10.1038/s41467-018-07552-9
50. Nault J.C., Datta S., Imbeaud S., Franconi A., Mallet M., Couchy G., et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat Genet. 2015;47(10):1187–93. DOI: 10.1038/ng.3389
51. Letouzé E., Shinde J., Renault V., Couchy G., Blanc J.F., Tubacher E., et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat Commun. 2017;8(1):1315. DOI: 10.1038/s41467-017-01358-x
52. Rebouissou S., Nault J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol. 2020;72(2):215–29. DOI: 10.1016/j.jhep.2019.08.017
53. Lachenmayer A., Alsinet C., Savic R., Cabellos L., Toffanin S., Hoshida Y., et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin Cancer Res. 2012;18(18):4997–5007. DOI: 10.1158/1078-0432.CCR-11-2322
54. Ruiz de Galarreta M., Bresnahan E., Molina-Sánchez P., Lindblad K.E., Maier B., Sia D., et al. β-Catenin activation promotes immune escape and resistance to Anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9(8):1124–41. DOI: 10.1158/2159-8290.CD-19-0074
55. Renehan A.G., Tyson M., Egger M., Heller R.F., Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371(9612):569–78. DOI: 10.1016/S0140-6736(08)60269-X
56. Sutti S., Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol. 2020;17(2):81–92. DOI: 10.1038/s41575-019-0210-2
57. Nakagawa H., Umemura A., Taniguchi K., Font-Burgada J., Dhar D., Ogata H., et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell. 2014;26(3):331–43. DOI: 10.1016/j.ccr.2014.07.001
58. Nishida N., Yada N., Hagiwara S., Sakurai T., Kitano M., Kudo M. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2016;31(9):1646–53. DOI: 10.1111/jgh.13318
59. Guri Y., Colombi M., Dazert E., Hindupur S.K., Roszik J., Moes S., et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell. 2017;32(6):807–23.e12. DOI: 10.1016/j.ccell.2017.11.011
60. Liu D., Wong C.C., Fu L., Chen H., Zhao L., Li C., et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci Transl Med. 2018;10(437):eaap9840. DOI: 10.1126/scitranslmed.aap9840
61. Umemura A., He F., Taniguchi K., Nakagawa H., Yamachika S., Font-Burgada J., et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell. 2016;29(6):935–48. DOI: 10.1016/j.ccell.2016.04.006
62. Grohmann M., Wiede F., Dodd G.T., Gurzov E.N., Ooi G.J., Butt T., et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell. 2018;175(5):1289–306.e20. DOI: 10.1016/j.cell.2018.09.053
63. Henderson J.M., Zhang H.E., Polak N., Gorrell M.D. Hepatocellular carcinoma: Mouse models and the potential roles of proteases. Cancer Lett. 2017;387:106–13. DOI: 10.1016/j.canlet.2016.03.047
64. Negro F. Natural history of NASH and HCC. Liver Int. 2020;40 Suppl 1:72–6. DOI: 10.1111/liv.14362
65. Rudalska R., Dauch D., Longerich T., McJunkin K., Wuestefeld T., Kang T.W., et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat Med. 2014;20(10):1138–46. DOI: 10.1038/nm.3679
66. Martinez-Quetglas I., Pinyol R., Dauch D., Torrecilla S., Tovar V., Moeini A., et al. IGF2 Is Up-regulated by epigenetic mechanisms in hepatocellular carcinomas and is an actionable oncogene product in experimental models. Gastroenterology. 2016;151(6):1192–205. DOI: 10.1053/j.gastro.2016.09.001
67. Doudna J.A., Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. DOI: 10.1126/science.1258096
68. Cook N., Jodrell D.I., Tuveson D.A. Predictive in vivo animal models and translation to clinical trials. Drug Discov Today. 2012;17(5–6):253–60. DOI: 10.1016/j.drudis.2012.02.003
69. Singh M., Ferrara N. Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat Biotechnol. 2012;30:648–57. DOI: 10.1038/nbt.2286
70. Newell P., Villanueva A., Friedman S.L., Koike K., Llovet J.M. Experimental models of hepatocellular carcinoma. J Hepatol. 2008;48(5):858–79. DOI: 10.1016/j.jhep.2008.01.008
71. Bresnahan E., Ramadori P., Heikenwalder M., Zender L., Lujambio A. Novel patient-derived preclinical models of liver cancer. J Hepatol. 2020;72(2):239–49. DOI: 10.1016/j.jhep.2019.09.028
72. Moriya K., Fujie H., Shintani Y., Yotsuyanagi H., Tsutsumi T., Ishibashi K., et al. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4(9):1065–7. DOI: 10.1038/2053
73. Hagel M., Miduturu C., Sheets M., Rubin N., Weng W., Stransky N., et al. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov. 2015;5(4):424–37. DOI: 10.1158/2159-8290.CD-14-1029
74. Day C.P., Merlino G., Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015;163(1):39–53. DOI: 10.1016/j.cell.2015.08.068
75. Jayson G., Harris J. How participants in cancer trials are chosen: ethics and conflicting interests. Nat Rev Cancer. 2006;6(4):330–6. DOI: 10.1038/nrc1842
76. Febbraio M.A., Reibe S., Shalapour S., Ooi G.J., Watt M.J., Karin M. Preclinical models for studying NASH-driven HCC: How useful are they? Cell Metab. 2019;29(1):18–26. DOI: 10.1016/j.cmet.2018.10.012
77. Sharpless N.E., Depinho R.A. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. 2006;5(9):741–54. DOI: 10.1038/nrd2110
78. Wolf M.J., Adili A., Piotrowitz K., Abdullah Z., Boege Y., Stemmer K., et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell. 2014;26(4):549–64. DOI: 10.1016/j.ccell.2014.09.003
79. Ma C., Kesarwala A.H., Eggert T., Medina-Echeverz J., Kleiner D.E., Jin P., et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531(7593):253–7. DOI: 10.1038/nature16969
80. Malehmir M., Pfister D., Gallage S., Szydlowska M., Inverso D., Kotsiliti E., et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019;25(4):641–55. DOI: 10.1038/s41591-019-0379-5
81. Ringelhan M., Pfister D., O’Connor T., Pikarsky E., Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19(3):222–32. DOI: 10.1038/s41590-018-0044-z
82. Wada Y., Nakashima O., Kutami R., Yamamoto O., Kojiro M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology. 1998;27(2):407–14. DOI: 10.1002/hep.510270214
83. Yuan D., Huang S., Berger E., Liu L., Gross N., Heinzmann F., et al. Kupffer cell-derived Tnf triggers cholangiocellular tumorigenesis through JNK due to chronic mitochondrial dysfunction and ROS. Cancer Cell. 2017;31(6):771–89.e6. DOI: 10.1016/j.ccell.2017.05.006
84. Crispe I.N. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–63. DOI: 10.1146/annurev.immunol.021908.132629
85. Horwitz E., Stein I., Andreozzi M., Nemeth J., Shoham A., Pappo O., et al. Human and mouse VEGFA-amplified hepatocellular carcinomas are highly sensitive to sorafenib treatment. Cancer Discov. 2014;4(6):730–43. DOI: 10.1158/2159-8290.CD-13-0782
86. Finn R.S., Ikeda M., Zhu A.X., Sung M.W., Baron A.D., Kudo M., et al. Phase Ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J Clin Oncol. 2020;38(26):2960–70. DOI: 10.1200/JCO.20.00808
87. Hou J., Zhang H., Sun B., Karin M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J Hepatol. 2020;72(1):167–82. DOI: 10.1016/j.jhep.2019.08.014
88. Hoshida Y., Villanueva A., Kobayashi M., Peix J., Chiang D.Y., Camargo A., et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359(19):1995–2004. DOI: 10.1056/NEJMoa0804525
89. Shalapour S., Lin X.J., Bastian I.N., Brain J., Burt A.D., Aksenov A.A., et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. 2017;551(7680):340–5. DOI: 10.1038/nature24302
90. Kang T.W., Yevsa T., Woller N., Hoenicke L., Wuestefeld T., Dauch D., et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–51. DOI: 10.1038/nature10599
91. Flecken T., Schmidt N., Hild S., Gostick E., Drognitz O., Zeiser R., et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59(4):1415–26. DOI: 10.1002/hep.26731
92. Zheng C., Zheng L., Yoo J.K., Guo H., Zhang Y., Guo X., et al. Landscape of infiltrating T cells in liver cancer revealed by singlecell sequencing. Cell. 2017;169(7):1342–56.e16. DOI: 10.1016/j.cell.2017.05.035
93. Langhans B., Nischalke H.D., Krämer B., Dold L., Lutz P., Mohr R., et al. Role of regulatory T cells and checkpoint inhibition in hepatocellular carcinoma. Cancer Immunol Immunother. 2019;68(12):2055–66. DOI: 10.1007/s00262-019-02427-4
94. Bruno T.C. New predictors for immunotherapy responses sharpen our view of the tumour microenvironment. Nature. 2020;577(7791):474–6. DOI: 10.1038/d41586-019-03943-0
95. Schneider C., Teufel A., Yevsa T., Staib F., Hohmeyer A., Walenda G., et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut. 2012;61(12):1733–43. DOI: 10.1136/gutjnl-2011-301116
96. Sautès-Fridman C., Petitprez F., Calderaro J., Fridman W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19(6):307–25. DOI: 10.1038/s41568-019-0144-6
97. Calderaro J., Petitprez F., Becht E., Laurent A., Hirsch T.Z., Rousseau B., et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol. 2019;70(1):58–65. DOI: 10.1016/j.jhep.2018.09.003
98. Finkin S., Yuan D., Stein I., Taniguchi K., Weber A., Unger K., et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol. 2015;16(12):1235–44. DOI: 10.1038/ni.3290
99. Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. DOI: 10.1038/nrgastro.2017.38
100. Higashi T., Friedman S.L., Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017;121:27–42. DOI: 10.1016/j.addr.2017.05.007
101. Dapito D.H., Mencin A., Gwak G.Y., Pradere J.P., Jang M.K., Mederacke I., et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21(4):504–16. DOI: 10.1016/j.ccr.2012.02.007
102. Ma C., Han M., Heinrich B., Fu Q., Zhang Q., Sandhu M., et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391):eaan5931. DOI: 10.1126/science.aan5931
103. Hoshida Y., Villanueva A., Sangiovanni A., Sole M., Hur C., Andersson K.L., et al. Prognostic gene expression signature for patients with hepatitis C-related early-stage cirrhosis. Gastroenterology. 2013;144(5):1024–30. DOI: 10.1053/j.gastro.2013.01.021
104. Budhu A., Forgues M., Ye Q.H., Jia H.L., He P., Zanetti K.A., et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10(2):99–111. DOI: 10.1016/j.ccr.2006.06.016
105. Moeini A., Torrecilla S., Tovar V., Montironi C., Andreu-Oller C., Peix J., et al. An immune gene expression signature associated with development of human hepatocellular carcinoma identifies mice that respond to chemopreventive agents. Gastroenterology. 2019;157(5):1383–97.e11. DOI: 10.1053/j.gastro.2019.07.028
к.м.н., доцент, кафедра онкологии с курсами онкологии и патологической анатомии ИДПО, отдел химиотерапии
Уфа
к.м.н., отдел противоопухолевой лекарственной терапии
Уфа
к.м.н., хирургическое отделение № 6
Уфа
д.м.н., профессор, кафедра онкологии с курсами онкологии и патологической анатомии ИДПО
Уфа
к.м.н., доцент, кафедра биологической химии
Уфа
к.м.н., хирургическое отделение № 1, orcid.org/0000-0003-
1438-2006
Уфа
отделение противоопухолевой лекарственной терапии № 1
Уфа
отделение амбулаторной противоопухолевой лекарственной
терапии
Уфа
кафедра онкологии с курсами онкологии и патологической анатомии ИДПО
Уфа
Меньшиков К.В., Султанбаев А.В., Мусин Ш.И., Рахматуллина И.Р., Меньшикова И.А., Абдеев Р.Р., Султанбаева Н.И., Попова Е.В., Серебренников Г.А. Гепатоцеллюлярная карцинома: этиологические факторы и механизмы развития. Обзор литературы. Креативная хирургия и онкология. 2022;12(2):139-150. https://doi.org/10.24060/2076-3093-2022-12-2-139-150
Menshikov K.V., Sultanbaev A.V., Musin Sh.I., Rakhmatullina I.R., Menshikova I.A., Abdeev R.R., Sultanbaeva N.I., Popova E.V., Serebrennikov G.A. Hepatocellular Carcinoma: Aetiology and Mechanisms of Development. A Literature Review. Creative surgery and oncology. 2022;12(2):139-150. (In Russ.) https://doi.org/10.24060/2076-3093-2022-12-2-139-150
Федеральное государственное бюджетное образовательное учреждение высшего образования «Башкирский государственный медицинский университет» Министерства здравоохранения Российской Федерации
450008, Республика Башкортостан, г. Уфа, ул. Пушкина, д. 96, корп. 98
Тел./факс: +7 (347) 273-56 -97
E-mail: csurgonco@bashgmu.ru






























