




Содержание
Перейти к:
https://doi.org/10.24060/2076-3093-2024-14-2-116-126
Перейти к:
Введение. Рак шейки матки (РШМ) остается наиболее распространенным раком у женщин во всем мире, и до сих пор для диагностики и прогнозирования РШМ отсутствуют эффективные и специфические биомаркеры. В последние годы большое внимание привлекло изучение потенциала циркулярных РНК (циркРНК) как новых диагностических, прогностических и терапевтических инструментов. В текущем исследовании мы провели углубленное биоинформатическое исследование по изучению регуляторной сети «циркРНК — микроРНК (миРНК)- матричной РНК (мРНК)», чтобы выявить важные молекулярные процессы и биологические пути, предположительно связанные с РШМ. Материалы и методы. В ходе исследования были собраны данные об экспрессии циркРНК (GSE102686), миРНК (GSE30656) и мРНК генов-мишеней (GSE9750), основанные на базе данных «Омнибус экспрессии генов» (англ. Gene Expression Omnibus, или GEO), в образцах плоскоклеточного рака шейки матки и нормального плоского эпителия шейки матки, разделив их на исследуемую и контрольную группы. Для более глубокого понимания функции циркРНК для их генов-мишеней был проведен анализ белок-белкового взаимодействия (PPI), Генной онтологии (GO — Gene Ontology) и анализ Киотской энциклопедии генов и геномов (KEGG). Результаты. В отношении РШМ было выявлено в общей сложности 105 дифференциально экспрессируемых циркулярных РНК (ДЭЦ), 144 дифференциально экспрессируемых микроРНК (ДЭМ) и 539 дифференциально экспрессируемых генов-мишеней (ДЭГ). Одновременно анализ функционального обогащения GO и путей KEGG проводился для ДЭГ. Впоследствии благодаря базам данных по поиску циркРНК, миРНК и мРНК генов-мишеней, а также сетевому анализу PPI и функциональному обогащению мы обнаружили 3 ДЭЦ со значительно более высоким уровнем экспрессии (hsa_circ_0000745, hsa_circ_0084927 и hsa_circ_0002762), 6 ДЭМ с пониженным уровнем экспрессии (hsa-miR-145, hsa-miR-876-3p, hsa-miR-1229, hsa-miR-182, hsa-miR520h и hsa-miR-1252) и 9 ключевых генов, таких как ANGPT2, COL11A1, MEST, KIF20A, CLN6, FNDC3B, USP18, DLGAP5 и CXCL9, что позволяет предположить их потенциально значительную роль при РШМ. Заключение. Понимание регуляторной сети «циркРНК — миРНК — мРНК» имеет большое значение в понимании онкогенеза РШМ, а также обнаружении новых циркРНК как главных регуляторных молекул в данной сети — это новое направление в диагностике и таргетной терапии РШМ.
Беглярзаде С.А., Тамразов Р.И., Мусаев Э.Р., Вонг Ч. Профиль экспрессии циркулярных РНК при раке шейки матки и построение регуляторной сети «циркулярные РНК — микроРНК — матричные РНК». Креативная хирургия и онкология. 2024;14(2):116-126. https://doi.org/10.24060/2076-3093-2024-14-2-116-126
Begliarzade S.A., Tamrazov R.I., Musaev E.R., Wang C. Circular RNA Expression Profile in Cervical Cancer and Construction of the Circular RNA‑MicroRNA‑Messenger RNA Regulatory Network. Creative surgery and oncology. 2024;14(2):116-126. (In Russ.) https://doi.org/10.24060/2076-3093-2024-14-2-116-126
Рак шейки матки (РШМ) является третьим по распространенности видом рака среди женщин во всем мире, только в 2020 году было зарегистрировано более 600 000 новых случаев [1]. Плоскоклеточный рак шейки матки составляет более 85 % всех случаев. При этом 5-летняя выживаемость при локализованном РШМ составляет около 91,5 %, а после метастазирования 5-летняя выживаемость снижается практически до 16,5 % [2]. Удовлетворительный прогноз на ранних стадиях РШМ можно объяснить недавними достижениями в традиционных стратегиях лечения. Однако примерно у двух третей пациенток РШМ диагностируется на поздней стадии из-за отсутствия практических диагностических биомаркеров, что делает стандартные стратегии лечения неэффективными [1–3]. Поэтому крайне важно изучить новые методы диагностики и терапии, опираясь на молекулярно-генетические особенности РШМ.
Полногеномные исследования с использованием микрочипов и секвенирования нового поколения (NGS) выявили глобальные эпигеномные изменения, ответственные за онкогенез, и предоставили возможность перевести эти молекулярные изменения в качестве биомаркеров и терапевтических мишеней у пациентов с РШМ. Тем не менее результаты полногеномного исследования потребовали перекрестной проверки, поскольку выявленные молекулы могут быть противоречивыми из-за: 1) гетерогенности опухоли, 2) используемых методик обнаружения, 3) типов и источников образцов и 4) алгоритмов, используемых для анализа данных [4][5]. Следовательно, повторный анализ больших данных может дать новое представление о регуляторных факторах, молекулярных механизмах и сигнальных путях, которые могут изменяться при РШМ. Предыдущие анализы in silico показали, что повторный анализ больших данных из базе данных «Омнибус экспрессии генов» (англ. Gene Expression Omnibus, или GEO) может выявить надежные диагностические и прогностические маркеры, а также терапевтические мишени при онкологических заболеваниях [6–8].
Циркулярные РНК (циркРНК) представляют собой класс некодирующих РНК (нРНК) с кольцевой конформацией, образующихся из пре-матричной РНК (мРНК) путем обратного сплайсинга. В отличие от линейных РНК, циркРНК более стабильны и устойчивы к расщеплению ферментами. Некоторые из циркРНК более распространены, чем их линейные транскрипты. Кроме того, циркРНК присутствуют в различных геномах млекопитающих [9][10]. Недавние исследования показали, что циркРНК играют все более важную роль в развитии многих заболеваний, и обнаружено, что аберрантная экспрессия циркРНК тесно связана с биогенезом и прогрессированием опухолей [11][12]. Несмотря на многочисленные функции циркРНК при опухолях человека, еще многое предстоит узнать о функциях и механизмах циркРНК, участвующих в онкогенезе РШМ, чтобы эффективно и безопасно транслировать фундаментальные знания в клиническую практику.
В этом исследовании мы получили образцы профиля экспрессии циркРНК, микроРНК (миРНК) и мРНК из базы данных GEO для пациентов с РШМ. С помощью языка программирования R были идентифицированы дифференциально экспрессируемые циркулярные РНК (ДЭЦ), дифференциально экспрессируемые микроРНК (ДЭМ) и дифференциально экспрессируемые гены-мишени (ДЭГ). Кроме того, были идентифицированы цели прогнозирования «циркРНК — миРНК» и «миРНК — мРНК». Затем путем их слияния была построена регуляторная сеть «циркРНК — миРНК — мРНК». Популярные методы обогащенного анализа, такие как анализ Белок-белкового взаимодействия (англ. Protein-Protein Interaction, или PPI), Онтология генов (англ. Gene Ontology, или GO) и анализ Киотской энциклопедии генов и геномов (англ. Kyoto Encyclopedia of Genes and Genomes, или KEGG), использовались для прогнозирования потенциальных сигнальных путей и регуляторных механизмов в онкогенезе РШМ. Это исследование предлагает новые диагностические и терапевтические инструменты, а также предпосылки для будущих исследований, которые могут улучшить наше понимание возможных молекулярных процессов РШМ.
В настоящем исследовании использовали ключевые слова «рак шейки матки или РШМ» (ключевые слова исследования), «циркулярные РНК или циркРНК» (тип исследования), «Home Sapiens» (организм) и «опухолевая ткань» (название атрибута) для извлечения данных из базы данных Национального центра биотехнологической информации по экспрессии генов (англ. National Center for Biotechnology Information, или NCBI) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc =GSE102686; доступ в декабре 2023 г.). Данные дифференциально экспрессируемых циркулярных РНК (ДЭЦ), дифференциально экспрессируемых микроРНК (ДЭМ) и дифференциально экспрессируемых генов-мишеней (ДЭГ) образцов РШМ и нормальных тканей были загружены из базы данных GEO, набора данных GSE102686, GSE30656 и GSE9750, которые основаны на микрочипе V1 GPL19978 Agilent-069978 Arraystar Human CircRNA (Agilent Technologies Inc., MD), GPL6955 Agilent-016436 Human miRNA Microarray 1.0 и GPL96[ HG-U133A] Affymetrix Human Genome U133A Array. Эти образцы включали 5 образцов плоскоклеточного рака и 5 образцов нормального эпителия шейки матки из базы данных GSE102686, 10 образцов плоскоклеточного рака и 10 образцов нормального эпителия шейки матки из базы данных GSE30656 и 40 образцов плоскоклеточного рака и 24 образца нормального эпителия шейки матки из базы данных GSE9750 (табл. 1). ДЭЦ, ДЭМ и ДЭГ между РШМ и нормальной тканью проверяли с помощью инструмента GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/?acc=GSE102686). Log2-кратное изменение |log2FC| > 1,0 и скорректированное значение p < 0,05 были определены в качестве отправной точки для выбора ДЭЦ, ДЭМ и ДЭГ.
GSE | Платформа | Образцы | Исследуемая группа, n | Контрольная группа, n | Молекула |
GSE102686 | GPL19978 | Плоскоклеточный рак и образцы плоского эпителия шейки матки | 5 | 5 | Циркулярные РНК (циркРНК) |
GSE30656 | GPL6955 | Плоскоклеточный рак и образцы плоского эпителия шейки матки | 10 | 10 | МикроРНК (миРНК) |
GSE9750 | GPL96 | Плоскоклеточный рак и образцы плоского эпителия шейки матки | 40 | 24 | Матричная РНК (мРНК) гена |
Таблица 1. Подробная информация о данных Gene Expression Omnibus (GEO)
Table 1. Gene Expression Omnibus (GEO) data details
Мы использовали базу данных circBase (http://www.circbase.org/) для анализа полученных ДЭЦ. CircBase — это база данных, связанная с поиском существующих циркРНК, в которой указывается краткая информация о циркРНК, такая как последовательность, ген и расположение в геноме [13]. Затем мы определили сеть взаимодействия «циркРНК — миРНК», используя базу данных circBank (http://www.circbank.cn/index.html) и базу данных CircInteractome (https://circinteractome.nia.nih.gov/index.html), где в сеть взаимодействия были включены миРНК с наивысшим баллом, контекст + процентиль оценки > 90 (англ. Context + percentile score) [13][14]. Далее три алгоритма прогнозирования мишеней, такие как TargetScan (https://www.targetscan.org/vert_80/), miRDB (https://mirdb.org/custom.html) и RNA22 (https://cm.jefferson.edu/rna22/) были использованы для предсказания потенциальной целевой взаимосвязи между выбранными миРНК и областями 3′-нетранслируемой области (3′-НТО, англ. 3′-untranslated region, 3′-UTR) 3′-НТО мРНК генов-мишеней [15]. Высокая степень достоверности присваивается, когда совокупная взвешенная оценка контекста (англ. Cumulative weighted context++ score или «CWCS»), определенная TargetScan, равна -0,4 или ниже. Такие оценки предсказывают, что миРНК подавляет конкретную мишень мРНК гена по меньшей мере на 25 % относительно нормального уровня. Умеренная достоверность присваивается, когда CWCS находится в диапазоне от -0,2 до -0,4. Такие оценки предсказывают, что миРНК подавляет конкретную мишень мРНК на 13–25 % по сравнению с их нормальными уровнями. По опыту многих исследователей, прогнозируемая мРНК как мишень с рейтингом прогнозирования > 80, контекст ++ процентиль оценки (англ. Context ++ percentile score) из базы данных miRDB имеет подтверждающие доказательства о практической взаимосвязями между миРНК и 3′-НТО мРНК генов-мишеней.
Потенциальные функции ДЭЦ были дополнительно исследованы путем изучения мРНК генов-мишеней в регуляторной сети. Обогащение целевых мРНК по путям GO и KEGG анализировали и визуализировали с использованием программного обеспечения База данных для аннотации, визуализации и интегрированного обнаружения (англ. The Database for Annotation, Visualization and Integrated Discovery, или DAVID). GO в основном используется для анализа функций мРНК, обогащенных клеточным компонентом (КК), молекулярной функцией (МФ) и биологическим процессом (БП). KEGG использовалась для анализа функций мРНК генов-мишеней, участвующих в нескольких сигнальных путях.
Статистический анализ проводили с использованием программного обеспечения IBM SPSS (версия 16.0) и Graphpad Prism (версия 8.0). Для сравнения различных групп использовался независимый t-критерий или критерий хи-квадрат. Вероятность p < 0,05 (*) или p < 0,001 (**) считалась статистически значимой.
В настоящем исследовании мы проанализировали наборы данных GSE102686, GSE30656 и GSE9750 из базы данных GEO с помощью инструмента GEO2R и обнаружили ДЭЦ при сравнении 5 образцов плоскоклеточного рака и 5 образцов нормального плоского эпителия шейки матки, ДЭМ при сравнении 10 образцов плоскоклеточного рака и 10 образцов нормального плоского эпителия шейки матки и ДЭГ при сравнении 40 образцов плоскоклеточного рака и 24 образцов нормального плоского эпителия шейки матки. Всего было обнаружено 105 ДЭЦ, 144 ДЭМ и 539 ДЭГ (p < 0,05 и p < 0,001). Среди 105 ДЭЦ 49 циркРНК имели повышенный уровень экспрессии и 56 циркРНК имели пониженный уровень экспрессии, среди 144 ДЭМ 27 миРНК имели повышенный уровень экспрессии и 117 миРНК имели пониженный уровень экспрессии, среди 539 ДЭГ 266 мРНК генов имели повышенный уровень экспрессии и 273 мРНК генов имели пониженный уровень экспрессии. Тепловые карты (heat maps) и Volcano plot диаграммы ДЭЦ, ДЭМ и ДЭГ в трех вышеуказанных наборах данных показаны на рисунках 1–3. Интерпретация изменения уровня экспрессии ДЭЦ, ДЭМ и ДЭГ основана на значениях log2FC > 1,0 (повышенный уровень экспрессии) и log2FC < -1,0 (пониженный уровень экспрессии).
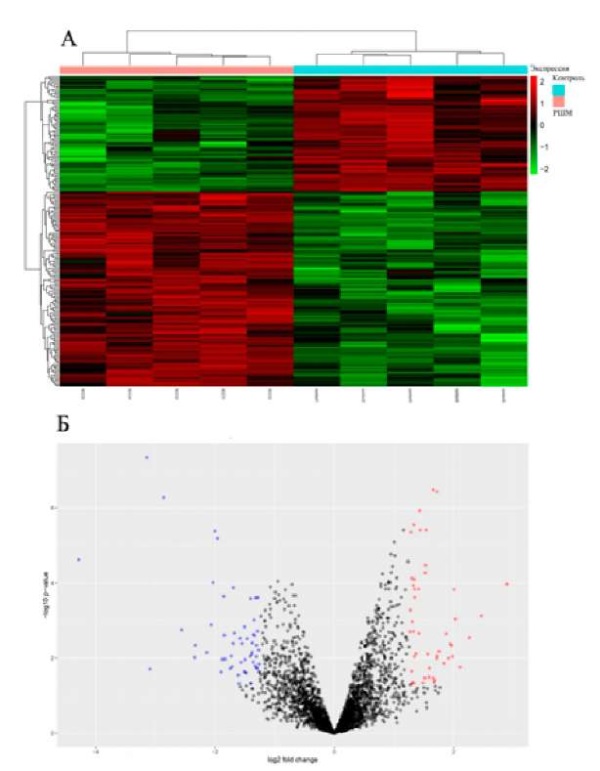
Рисунок 1. Профиль экспрессии и кластерный анализ циркулярных РНК (циркРНК) в образцах плоскоклеточного рака шейки матки (РШМ) и образцах нормального эпителия шейки матки (контрольная группа) из базы данных GSE102686. А — тепловая карта иллюстрирует значения экспрессии дифференциально экспрессируемых циркулярных РНК (ДЭЦ). Цветовая схема назначает розовый цвет образцам РШМ, а бирюзовый цвет — контрольной группе, эффективно отображая отличительные закономерности экспрессии между двумя группами, где красный цвет — это высокий уровень экспрессии, зеленый цвет — низкий уровень экспрессии циркРНК; Б — volcano plot диаграмма демонстрирует ДЭЦ, подчеркивая взаимосвязь между статистической значимостью и кратностью изменения. Красные точки указывают на увеличение уровня экспрессии, синие точки — снижение уровня экспрессии, а серые точки — отсутствие значимой дифференциальной экспрессии. Это визуальное представление кратко передает масштабы и значимость изменений экспрессии анализируемых циркРНК
Figure 1. Expression profile and cluster analysis of circular RNAs (circRNA) in cervical squamous cell carcinoma (cervical cancer) samples and cervical normal epithelium samples (control group) from the GSE102686 database. A — heat map illustrates the expression values of differentially expressed circular RNAs (DECs). The color scheme assigns pink to cervical cancer samples and turquoise to the control group, illustrating the distinctive expression patterns between the two groups, with red representing high-level expression and green representing low-level circRNA expression; B — volcano plot diagram demonstrates DECs, highlighting the relationship between statistical significance and fold change. Red dots indicate an increase in expression level, blue dots denote a decrease in expression level, and gray dots designate no significant differential expression. This visual representation summarizes the magnitude and significance of the expression changes of the circRNAs analyzed
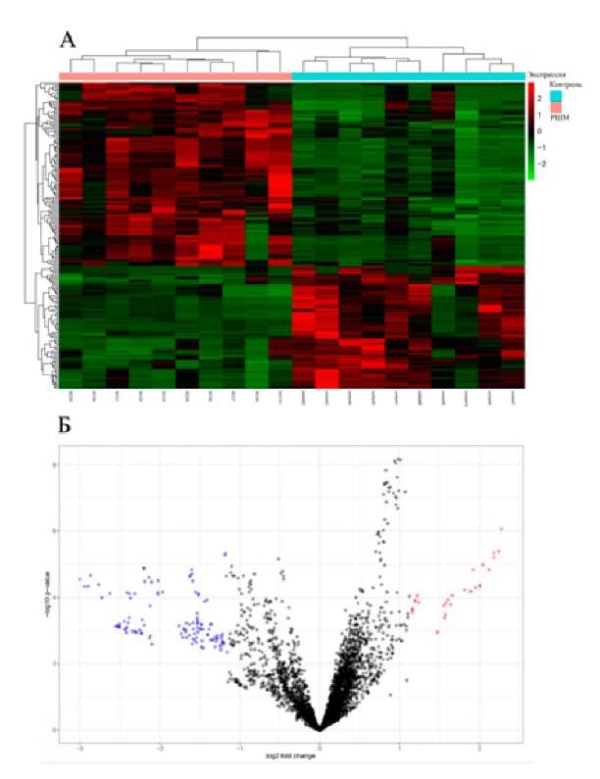
Рисунок 2. Профиль экспрессии и кластерный анализ микроРНК (миРНК) в образцах плоскоклеточного рака шейки матки (РШМ) и образцах нормального эпителия шейки матки (контрольная группа) из базы данных GSE30656. А — тепловая карта иллюстрирует значения экспрессии дифференциально экспрессируемых микроРНК (ДЭМ). Цветовая схема назначает розовый цвет образцам РШМ, а бирюзовый цвет — контрольной группе, эффективно отображая отличительные закономерности экспрессии между двумя группами, где красный цвет — это высокий уровень экспрессии, зеленый цвет — низкий уровень экспрессии миРНК; Б — volcano plot диаграмма демонстрирует ДЭМ, подчеркивая взаимосвязь между статистической значимостью и кратностью изменения. Красные точки указывают на увеличение уровня экспрессии, синие точки — снижение уровня экспрессии, а серые точки — отсутствие значимой дифференциальной экспрессии. Это визуальное представление кратко передает масштабы и значимость изменений экспрессии анализируемых миРНК
Figure 2. Expression profile and cluster analysis of microRNAs (miRNA) in cervical squamous cell carcinoma (cervical cancer) samples and cervical normal epithelium samples (control group) from the GSE30656 database. A — heat map illustrates the expression values of differentially expressed microRNAs (DEMs). The color scheme assigns pink to cervical cancer samples and turquoise to the control group, illustrating the distinctive expression patterns between the two groups, with red representing high-level expression and green representing low-level miRNA expression; B — volcano plot diagram demonstrates DEMs, highlighting the relationship between statistical significance and fold change. Red dots indicate an increase in expression level, blue dots denote a decrease in expression level, and gray dots designate no significant differential expression. This visual representation summarizes the magnitude and significance of the expression changes of the miRNAs analyzed
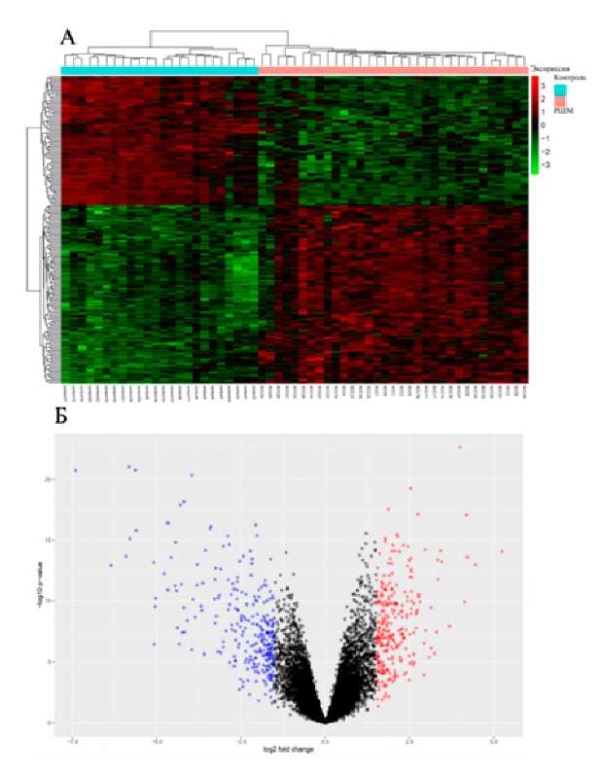
Рисунок 3. Профиль экспрессии и кластерный анализ генов-мишеней в образцах плоскоклеточного рака шейки матки (РШМ) и образцах нормального эпителия шейки матки (контрольная группа) из базы данных GSE9750. А — тепловая карта иллюстрирует значения экспрессии дифференциально экспрессируемых генов-мишеней (ДЭГ). Цветовая схема назначает розовый цвет образцам РШМ, а бирюзовый цвет — контрольной группе, эффективно отображая отличительные закономерности экспрессии между двумя группами, где красный цвет — это высокий уровень экспрессии, зеленый цвет — низкий уровень экспрессии генов-мишеней; Б — volcano plot диаграмма демонстрирует ДЭГ, подчеркивая взаимосвязь между статистической значимостью и кратностью изменения. Красные точки указывают на увеличение уровня экспрессии, синие точки — снижение уровня экспрессии, а серые точки — отсутствие значимой дифференциальной экспрессии. Это визуальное представление кратко передает масштабы и значимость изменений экспрессии анализируемых генов-мишеней
Figure 3. Expression profile and cluster analysis of target genes in cervical squamous cell carcinoma (cervical cancer) samples and cervical normal epithelium samples (control group) from the GSE9750 database. A — heat map illustrates the expression values of differentially expressed target genes (DEGs). The color scheme assigns pink to cervical cancer samples and turquoise to the control group, illustrating the distinctive expression patterns between the two groups, with red representing high-level expression and green representing low-level expression of target genes; B — volcano plot diagram demonstrates DEGs, highlighting the relationship between statistical significance and fold change. Red dots indicate an increase in expression level, blue dots denote a decrease in expression level, and gray dots designate no significant differential expression. This visual representation summarizes the magnitude and significance of the expression changes of the target genes analyzed
На основании установленных данных были выбраны 5 циркРНК с наиболее высоким уровнем экспрессии и 5 циркРНК с наиболее низким уровнем экспрессии. Этими 10 циркРНК были hsa_circ_0000745, hsa_circ_0084927, hsa_circ_0002762, hsa_circ_0075341, hsa_circ_0081672, hsa_circ_0031027, hsa_circ_0065898, hsa_circ_0046290, hsa_circ_0070190 и hsa_circ_0027821. Информация о кандидатных циркРНК приведена в таблице 2.
Название | Синоним | Тип | Локус | log2FC | Экспрессия |
hsa_circ_0000745 | hsa_circRNA_101996 | Экзогенная | chr17 | 2,888156 | Повышена |
hsa_circ_0084927 | hsa_circRNA_104651 | Экзогенная | сhr8 | 2,460979 | Повышена |
hsa_circ_0002762 | hsa_circRNA_101119 | Экзогенная | chr12 | 2,263959 | Повышена |
hsa_circ_0075341 | hsa_circRNA_104034 | Экзогенная | сhr5 | 2,100263 | Повышена |
hsa_circ_0081672 | hsa_circRNA_104443 | Экзогенная | chr7 | 2,026888 | Повышена |
hsa_circ_0031027 | hsa_circRNA_101308 | Экзогенная | chr13 | -4,28537 | Понижена |
hsa_circ_0065898 | hsa_circRNA_103384 | Экзогенная | сhr3 | -3,14143 | Понижена |
hsa_circ_0046290 | hsa_circRNA_102233 | Экзогенная | chr17 | -3,08777 | Понижена |
hsa_circ_0070190 | hsa_circRNA_103677 | Экзогенная | сhr4 | -2,86064 | Понижена |
hsa_circ_0027821 | hsa_circRNA_101120 | Экзогенная | chr12 | -2,55786 | Понижена |
Таблица 2. Кандидатные циркулярные РНК (циркРНК) при раке шейки матки (РШМ)
Table 2.Candidate circular RNAs (circRNA) in cervical cancer (CC)
Используя базу данных CircInteractome и circBank и учитывая параметры «контекст + процентиль оценки ≥ 90», мы идентифицировали 99 целевых миРНК для этих 10 циркРНК. Используя эти 99 предсказанных миРНК из баз данных для выбора миРНК мишеней, с пересечением с 144 ДЭМ, полученными из набора данных GSE30656, мы, наконец, получили 22 целевые миРНК с низким уровнем экспрессии, включая также те миРНК, которые имеют перекрестные связи из базы данных CircInteractome и circBank с кандидатными циркРНК: hsa-miR-145, hsa-miR-494, hsa-miR-876-3p, hsa-miR-182, hsa-miR-520g, hsa-miR-520h, hsa-miR-874, hsa-miR-1252, hsa-miR-579, hsa-miR-1256, hsa-miR-766, hsa-miR-146b-3p, hsa-miR-576-3p, hsa-miR-758, hsa-miR-513a-5p, hsa-miR-1229, hsa-miR-224, hsa-miR-338-5p, hsa-miR-567, hsa-miR-623, hsa-miR-370 и hsa-miR-638. 7 миРНК (hsa-miR-145, hsa-miR-494, hsa-miR-182, hsa-miR-224, hsa-miR-623, hsa-miR-370 и hsa-miR-638) из данных 22 миРНК имели совпадение циркРНК — миРНК взаимодействия как из базы данных CircInteractome и circBank, так и из набора данных GSE30656.
Далее три алгоритма прогнозирования целей, такие как TargetScan (https://www.targetscan.org/vert_80/), miRDB (https://mirdb.org/custom.html) и RNA22 (https://cm.jefferson.edu/rna22/), были использованы для предсказания потенциальной целевой взаимосвязи между найденными миРНК и 3′-НТО областями мРНК генов-мишеней. Используя информацию из этих баз данных о взаимодействиях предсказанных 22 миРНК с 3′-НТО областями мРНК генов-мишеней, с пересечением с 539 ДЭГ, полученными из набора данных GSE9750, мы получили 68 целевых генов с различным уровнем экспрессии: MPZL2, NAV3, ANGPT2, COL11A1, MEST, MXD1, TRPS1, KIF20A FNDC3B, BNIP3, EGR3, HBEGF, MAGEL2, CRISP3, DSG2, ZFP36, ENDOU, RRAGD, USP18, GJA1, RAPGEFL1, PKP1, IGF1, LPAR6, DKK2, GLTP, DLGAP5, CXCL9, PPP1R3C, CRABP2, RAB25, FUT8, STX11, KLK12, SERPINB13, CHL1, GREB1, DSG3, KAT2B, ARHGEF26, SNX10, PTGER3, SFRP1, PLK2, GOLM1, FN1, GPX3, CLN6, FOSB, SLC24A3, SERPINB2, PLOD2, MREG, ABCA8, CENPF, ID4, MECOM, SIX1, RRM2, PELI1, ELL2, SLC16A6, MEIS2, KLF4, NEK2, TMPRSS11E, KRT14 и EVPL.
Мы объединили пары «циркРНК — миРНК» и «миРНК — мРНК», чтобы реконструировать регуляторную сеть «циркРНК — миРНК — мРНК» при РШМ. Регуляторная сеть «циркРНК — миРНК — мРНК», или сеть PPI, была визуализирована с использованием программы Cytoscape (версия 3.10.1). Известно, что циркРНК играют роль в экспрессии генов, поскольку они могут частично ингибировать активность микроРНК. Они связываются с микроРНК, как губка, и действуют как конкурирующая эндогенная РНК, регулируя функцию целевой микроРНК, тем самым косвенно воздействуя на уровни мРНК. Поэтому, учитывая вышеизложенные результаты, мы пришли к окончательному выбору целевых циркРНК и их миРНК с мРНК генов-мишенями. В частности, были выбраны 3 циркРНК из 10 кандидатных (hsa_circ_0000745, hsa_circ_0084927 и hsa_circ_0002762) с повышенным уровнем экспрессии, 6 миРНК из 22 кандидатных (hsa-miR-145, hsa-miR-876-3p, hsa-miR-1229, hsa-miR-182, hsa-miR-520h и hsa-miR-1252) с пониженным уровнем экспрессии и 9 генов из 68 кандидатных (ANGPT2, COL11A1, MEST, KIF20A, CLN6, FNDC3B, USP18, DLGAP5 и CXCL9) с повышенным уровнем экспрессии. Предварительная и окончательная регуляторные сети «циркРНК — миРНК — мРНК» представлена на рисунках 4 и 5. Эти взаимодействия между циркРНК, миРНК и мРНК генов могут дать новое понимание механизма, лежащего в основе онкогенеза РШМ и внеклеточных коммуникаций через передачу циркулирующих РНК молекул в составе ВВ.
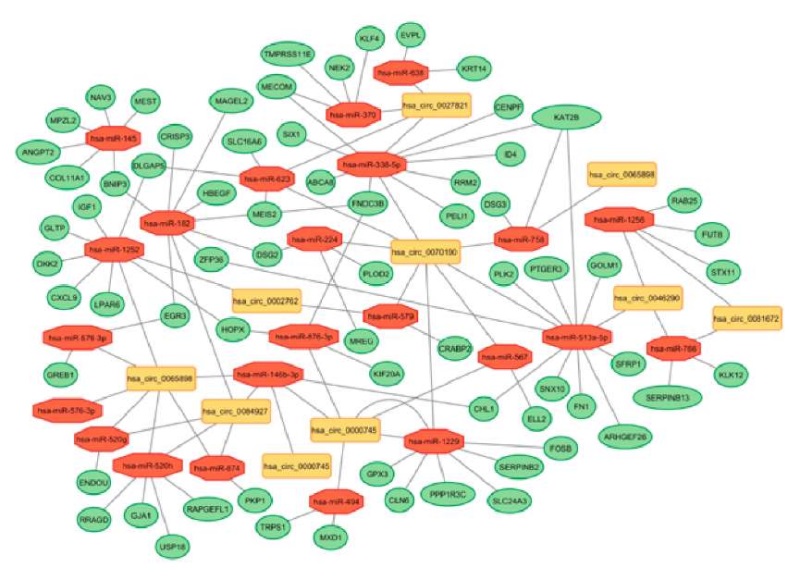
Рисунок 4. Анализ предварительной регуляторной сети взаимодействия циркулярных РНК (циркРНК) — микроРНК (миРНК) — матричной РНК (мРНК) генов-мишеней при раке шейки матки (РШМ). Эта визуализация дает представление о сложной сети взаимодействий между идентифицированными циркРНК, миРНК и генами-мишенями, обеспечивая всесторонний обзор их регуляторных взаимоотношений в контексте онкогенеза РШМ. Желтый цвет — циркРНК; красный цвет — миРНК; зеленый цвет — гены-мишени
Figure 4. Analysis of the preliminary circRNA-miRNA-mRNA regulatory network of target genes in cervical cancer (CC). This imaging provides insight into the complex network of interactions between the identified circRNAs, miRNAs, and target genes, providing a comprehensive overview of their regulatory relationships in the context of CC oncogenesis. Yellow — circRNA; red — miRNA; green — target genes
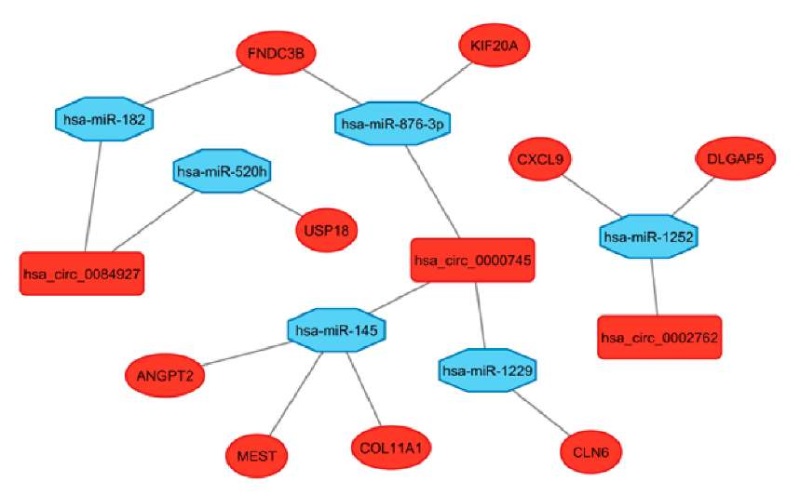
Рисунок 5. Анализ окончательной регуляторной сети взаимодействия циркулярных РНК (циркРНК) — микроРНК (миРНК) — матричной РНК (мРНК) генов-мишеней при раке шейки матки (РШМ). Красный цвет — повышенный уровень экспрессии, синий цвет — пониженный уровень экспрессии
Figure 5. Analysis of the final circRNA-miRNA-mRNA regulatory network of target genes in cervical cancer (CC). Red — increased expression level, blue — decreased expression level
Для обнаружения потенциальных функций hsa_circ_0000745, hsa_circ_0084927 и hsa_circ_0002762 использовали 9 ключевые генов, включая анализ обогащения GO и анализ пути KEGG. При анализе GO, включая БП, КК и МФ, мы получили 30 результатов для 9 ДЭГ (при БП были 8 ДЭГ из 9). При анализе пути KEGG мы получили 12 результатов для 4 ДЭГ. В таблице 3 показана полная информация каждого обнаруженного кластера GO и для путей KEGG. Количество — это количество генов, соответствующих базе данных путей, а % — процент совпадений генов среди общего числа генов в базе данных путей. Кратное обогащение (англ. Fold enrichment) определяется как процент генов в общем списке, принадлежащих к сигнальному пути, разделенный на соответствующий процент в фоновом режиме. Кратное обогащение показывает, насколько сильно представлены гены определенного сигнального пути. Популяционное попадание (англ. Pop Hits) показывает, сколько генов имеет название функции в интересующем в списке генов, а общее количество популяций (англ. Pop Total) показывает, сколько генов в общей популяции имеют это название функции в фоновом геноме (все гены интересующего вида в базе данных DAVID). Значения p были проанализированы с использованием точного показателя Фишера. Данные считались значимыми, если p < 0,05 или p < 0,001.
Гены | Количество | KEGG ID | Термины | % | Pop Hits | Pop Total | Кратное обогащение | Значение p |
CXCL9 | 1 | hsa04060 | Взаимодействие цитокинов и цитокиновых рецепторов | 11,1 | 297 | 8644 | 7,27 | 0,03 |
CXCL9 | 1 | hsa04061 | Взаимодействие вирусного белка с цитокином и цитокиновым рецептором | 11,1 | 100 | 8644 | 21,61 | 0,03 |
CXCL9 | 1 | hsa04062 | Сигнальный путь хемокинов | 11,1 | 192 | 8644 | 11,25 | 0,024 |
CXCL9 | 1 | hsa04620 | Сигнальный путь Toll-подобного рецептора | 11,1 | 108 | 8644 | 20,0 | 0,011 |
COL11A1 | 1 | hsa04974 | Переваривание и абсорбция белков | 11,1 | 103 | 8644 | 20,98 | 0,038 |
ANGPT2 | 1 | hsa04014 | Сигнальный путь Ras | 11,1 | 236 | 8644 | 9,156 | 0,01 |
ANGPT2 | 1 | hsa04151 | Сигнальный путь PI3K-Akt | 11,1 | 359 | 8644 | 6,019 | < 0,001 |
ANGPT2 | 1 | hsa04015 | Сигнальный путь Rap1 | 11,1 | 210 | 8644 | 10,29 | 0,049 |
ANGPT2 | 1 | hsa05167 | Герпесвирусная инфекция, ассоциированная с саркомой Капоши | 11,1 | 194 | 8644 | 11,13 | 0,05 |
ANGPT2 | 1 | hsa04066 | Сигнальный путь HIF-1 | 11,1 | 109 | 8644 | 19,8 | < 0,001 |
ANGPT2 | 1 | hsa04010 | Сигнальный путь МАРК | 11,1 | 301 | 8644 | 7,17 | 0,012 |
KIF20A | 1 | hsa04814 | Моторные белки | 11,1 | 193 | 8644 | 11,19 | 0,041 |
Таблица 3. Анализ Киотской энциклопедии генов и геномов (KEGG)
Table 3. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis
Результаты GO анализа показали, что изменения БП 8 ДЭГ были значительно обогащены визуальном восприятием, преобразованием сигнала, негативной регуляцией сигнального пути, опосредованного интерфероном I типа, организацией митотического веретена, развитием мезодермы, транспортом белка, негативной регуляцией ангиогенеза, метаболическим процессом протеогликанов, регуляцией цитокинеза и регуляцией накопления липидов (рис. 6).
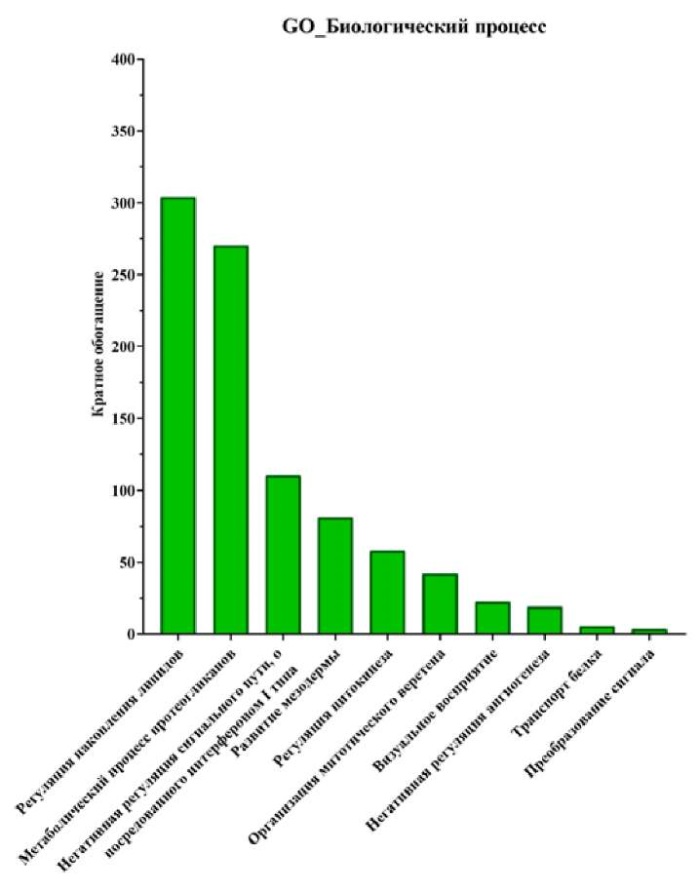
Рисунок 6. Анализ Онтологии генов (GO) для 8 дифференциально экспрессируемых генов-мишеней (ДЭГ) из 9, обогащенных клеточным компонентом биологическим процессом (БП)
Figure 6. Gene Ontology (GO) analysis for 8 differentially expressed target genes (DEGs) of 9, enriched in biological process
Изменения КК 9 ДЭГ касались преимущественно ядра, внеклеточного пространства, внеклеточной области, неотъемлемого компонента мембраны, эндоплазматического ретикулума, внутриклеточной мембраносвязанной органеллы, мембраны эндоплазматической сети, мембран, цитозоли и проекции клетки (рис. 7).
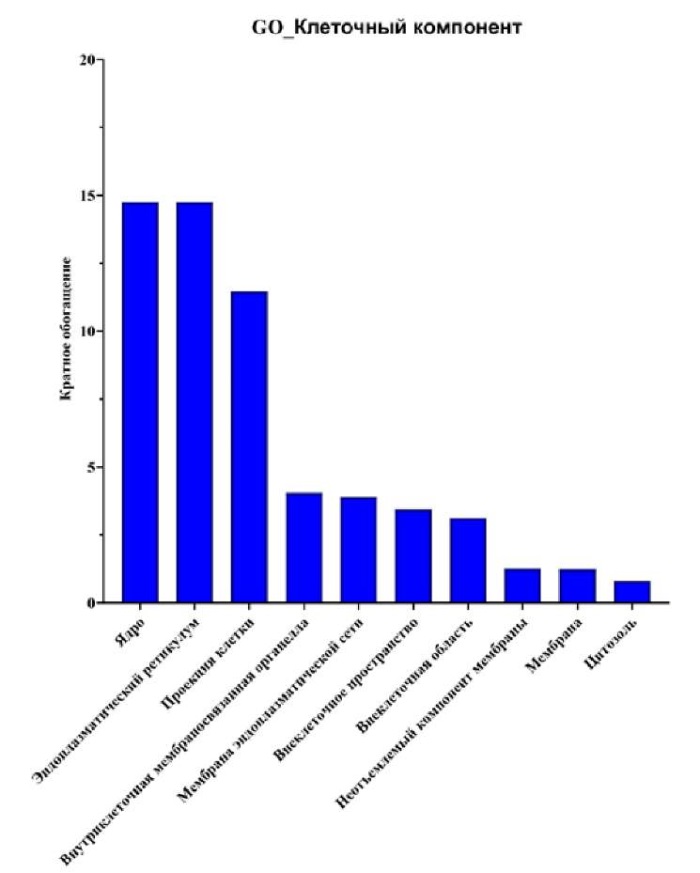
Рисунок 7. Анализ Онтологии генов (GO) для 9 дифференциально экспрессируемых генов-мишеней (ДЭГ) из 9, обогащенных клеточным компонентом (КК)
Figure 7. Gene Ontology (GO) analysis for 9 differentially expressed target genes (DEGs) of 9, enriched in cellular component
Изменения МФ в основном касались cвязывания белка, микротрубочек, ионов металлов, РНК, АТФ, протеинкиназ, рецепторной тирозинкиназы, а также гидролазной активности, структурных компонентов внеклеточного матрикса и активности гомодимеризации белка (рис. 8).
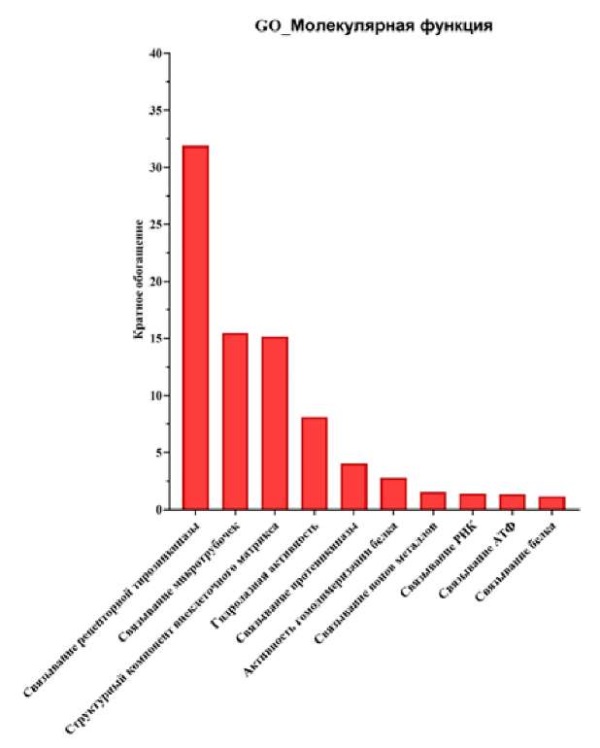
Рисунок 8. Анализ Онтологии генов (GO) для 9 дифференциально экспрессируемых генов-мишеней (ДЭГ) из 9, обогащенных молекулярной функцией (МФ)
Figure 8. Gene Ontology (GO) analysis for 9 differentially expressed target genes (DEGs) of 9, enriched in molecular function
Анализ пути KEGG показал, что 4 ДЭГ в основном были обогащены взаимодействием цитокинов и цитокиновых рецепторов, взаимодействием вирусного белка с цитокином и цитокиновым рецептором, сигнальными путями хемокинов, Toll-подобного рецептора, перевариванием и абсорбцией белков, сигнальными путями Ras, PI3K-Akt, Rap1, герпесвирусной инфекцией, ассоциированной с саркомой Капоши, сигнальными путями HIF-1, МАРК и моторными белками (рис. 9).
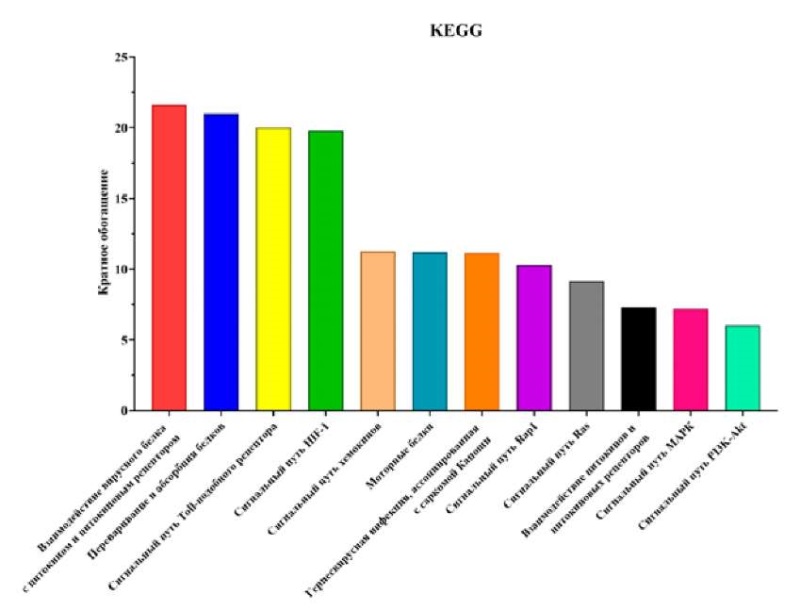
Рисунок 9. Анализ путей Киотской энциклопедии генов и геномов (KEGG) для 4 дифференциально экспрессируемых генов-мишеней (ДЭГ) из 9, обогащенных 12 ключевыми сигнальными путями
Figure 9. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for 4 differentially expressed target genes (DEGs) out of 9, enriched in 12 key signalling pathways
В сигнальных путях, связанных с опухолью, анализ KEGG показал, что ANGPT2 и CXCL9 включены в большинство сигнальных путей, в том числе в хорошо изученные сигнальные пути, участвующие в онкогенезе РШМ, сигнальные пути Ras, PI3K-Akt, Rap1, HIF-1, МАРК, Toll-подобного рецептора и воспалительного процесса.
РШМ — распространенная гинекологическая опухоль. По уровню заболеваемости и смертности среди женских злокачественных опухолей занимает четвертое место в мире, уступая только раку молочной железы (РМЖ), колоректальному раку и раку легких. РШМ является одним из видов рака, который можно предотвратить с помощью скрининга [1–3]. Благодаря широкой популярности скрининга заболеваемость РШМ снижается с каждым годом. Однако, поскольку ранний РШМ не имеет симптомов, многие пациентки на момент постановки диагноза уже находятся на средней и поздних стадиях заболевания. При этом методы лечения пациентов на средней и поздних стадиях ограничены, а терапевтический эффект лучевой и химиотерапии не является удовлетворительным [16]. Поэтому есть необходимость в поиске новых молекулярных биомаркеров и терапевтических мишеней. Широкое применение баз данных биоинформатики способствовало открытию новых диагностических и терапевтических инструментов при онкологических заболеваниях, в том числе РШМ [17].
Достижения в области аналитических методов и биоинформатики открывают широкий спектр для проведения практических фундаментальных исследований. Биоинформатический анализ теперь можно проводить на любом биологическом образце (ткани или биологические жидкости). Каждый тип образца представляет собой потенциальный источник диагностических и прогностических биомаркеров и потенциальных терапевтических мишеней. Результаты, полученные нами в данном исследовании с использованием баз данных GEO, позволили идентифицировать новые циркРНК и их регуляторные сети «циркРНК — миРНК — мРНК», которые могут участвовать в развитии и прогрессировании РШМ. В настоящем исследовании мы получили исходный набор данных микрочипирования РШМ GSE102686 и идентифицировали 105 ДЭЦ, где 49 циркРНК имели повышенный уровень экспрессии и 56 циркРНК — пониженный уровень экспрессии. Впоследствии были определены 3 ключевые циркРНК: hsa_circ_0000745, hsa_circ_0084927 и hsa_circ_0002762.
В предыдущем исследовании было обнаружено, что уровень экспрессии hsa_circ_0000745 в тканях рака желудка был связан с дифференцировкой опухоли [18]. Более того, авторы определили его потенциальную диагностическую ценность. Zhang и др. в совокупности указали, что ингибирование экспрессии hsa_circ_0000745 может подавлять рост опухоли и способствовать апоптозу клеток РМЖ, резистентных к лучевой терапии, по крайней мере частично, через регуляторную сеть miR-1236-3p-CBX8, подтверждая, что hsa_circ_0000745 может быть терапевтической мишенью для разработки таргетного препарата при РМЖ [19]. Кроме того, Jiao и др., продемонстрировали, что снижение экспрессии hsa_circ_0000745 ингибирует пролиферацию, миграцию и инвазию клеток РШМ за счет повышения экспрессии белка E-кадгерина in vitro [20]. На основании данных NGS и предварительной экспериментальной проверки исследование Chen и др., показало, что hsa_circ_0084927 значительно сверхэкспрессируется в тканях и клеточных линиях аденокарциномы толстой кишки [21]. Было выявлено, что hsa_circ_0084927 может способствовать пролиферации, миграции и инвазии опухолевых клеток. Кроме того, эффективно была сконструирована регуляторная has_circ_0084927/miR-106b-5p/VEGFA. Данная регуляторная сеть указывает на то, что hsa_circ_0084927 может влиять на прогноз посредством регуляции клеточного цикла, апоптоза и других сигнальных путей. Также Shi и др., показали, что hsa_circ_0084927 активируется при РШМ [22]. Высокая экспрессия hsa_circ_0084927 была связана со степенью злокачественности по классификации, утвержденной Международной федерацией акушеров и гинекологов (International Federation of Gynecology and Obstetrics, или FIGO), метастазами в лимфатические узлы и инвазией опухоли, что означает, что hsa_circ_0084927 может быть новым молекулярным биомаркером при РШМ. Снижение уровня экспрессии hsa_circ_0084927 уменьшало рост опухоли in vivo и вызывало остановку клеточного цикла, апоптоз, подавляло образование колоний, пролиферацию, инвазию и опухолевых миграцию клеток in vitro. В другой работе было продемонстрировано, что hsa_circ_0002762 был высоко экспрессирован в тканях и клетках РШМ [23]. Инактивация hsa_circ_0002762 уменьшала пролиферацию, миграцию и инвазию опухолевых клеток. Инактивация hsa_circ_0002762 подавляла пролиферацию, миграцию и инвазию клеток РШМ путем активации опухолевого супрессора miR-122-5p. Более того, ASF1B был геном-мишенью miR-122-5p. Дополнительно эксперименты на животных подтвердили противоопухолевый эффект инактивации hsa_circ_0002762.
Данные исследования показывают, что циркРНК представляет собой тип высокоэффективной конкурирующей эндогенной РНК (англ. competing endogenous RNAs, или ceRNAs). Они могут ингибировать связывание миРНК с 3′-НТО мРНК генов-мишеней и регулировать уровень экспрессии генов-мишеней, оказывая эффект «секвестрации» миРНК [9]. Чтобы определить, действуют ли вышеуказанные 3 цикРНК как ceRNAs при РШМ, с помощью базы данных GSE30656 и GSE9750, circBase, circBank, CircInteractome, TargetScan, miRDB и RNA22 были предсказаны ключевые миРНК и ключевые гены-мишени. Кроме того, благодаря сетевому анализу PPI и функциональному обогащению GO и анализу путей KEGG было идентифицировано 10 пар «циркРНК — миРНК — мРНК» (hsa_circ_0000745-hsa-miR-145-ANGPT2, hsa_circ_0000745-hsa-miR-145-COL11A1, hsa_circ_0000745-hsa-miR-145-MEST, hsa_circ_0000745-hsa-miR-876-3p-KIF20A, hsa_circ_0000745-hsa-miR-876-3p-FNDC3B, hsa_circ_0000745-hsa-miR-1229-CLN6, hsa_circ_0084927-hsa-miR-182-FNDC3B, hsa_circ_0084927-hsa-miR-520h-USP18, hsa_circ_0002762-hsa-miR-1252-DLGAP5 и hsa_circ_0002762-hsa-miR-1252-CXCL9). Было обнаружено, что hsa_circ_0000745, hsa_circ_0084927 и hsa_circ_0002762 активируются тканях и клетках РШМ и в базе данных GSE102686, и эти результаты согласуются с предыдущими результатами. Примечательно, что гены-мишени регулируют ряд сигнальных путей, которые учувствуют в прогрессировании РШМ, а именно сигнальные пути Ras, PI3K-Akt, Rap1, HIF-1, МАРК, Toll-подобного рецептора и сигнальные пути воспалительного процесса, и эти пути требуют дальнейшего изучения [24–32].
В данном исследовании мы продемонстрировали профиль экспрессии циркРНК в тканях РШМ и идентифицировали 3 ключевых циркРНК, дифференциально экспрессируемых между тканями РШМ и нормальным эпителием шейки матки. Мы также идентифицировали 6 ключевых миРНК и 9 мРНК генов-мишеней с дифференциальной экспрессией из общедоступной базы данных GEO. Затем была построена регуляторная сеть «циркРНК — миРНК — мРНК». Результаты этого исследования обеспечивают возможный молекулярный механизм онкогенеза РШМ. Дальнейшие и дополнительные экспериментальные исследования, раскрывающие функции этих циркРНК, которые участвуют в инвазии и метастазировании РШМ, откроют новую перспективу для диагностики, прогнозирования и лечения данной патологии.
1. Podwika S.E., Duska L.R. Top advances of the year: Cervical cancer. Cancer. 2023;129(5):657–63. DOI: 10.1002/cncr.34617
2. Robinson E.F., Darby J.P., Moulder J.K. Cervical cancer screening: missed opportunities in a one-track model. Int J Gynecol Cancer. 2023;33(4):646. DOI: 10.1136/ijgc-2023-004311
3. Sokale I.O., Thrift A.P., Montealegre J., Adekanmbi V., Chido-Amajuoyi O.G., Amuta A., et al. Geographic variation in late-stage cervical cancer diagnosis. JAMA Netw Open. 2023;6(11):e2343152. DOI: 10.1001/jamanetworkopen.2023.43152
4. Martínez-Rodríguez F., Limones-González J.E., Mendoza-Almanza B., Esparza-Ibarra E.L., Gallegos-Flores P.I., Ayala-Luján J.L., et al. Understanding cervical cancer through proteomics. Cells. 2021;10(8):1854. DOI: 10.3390/cells10081854
5. Elias M.H., Das S., Abdul Hamid N. Candidate genes and pathways in cervical cancer: a systematic review and integrated bioinformatic analysis. Cancers (Basel). 2023;15(3):853. DOI: 10.3390/cancers15030853
6. Wu B., Xi S. Bioinformatics analysis of differentially expressed genes and pathways in the development of cervical cancer. BMC Cancer. 2021;21(1):733. DOI: 10.1186/s12885-021-08412-4
7. Han Y.H., Ma D.Y., Lee S.J., Mao Y.Y., Sun S.Y., Jin M.H., et al. Bioinformatics analysis of novel targets for treating cervical cancer by immunotherapy based on immune escape. Cancer Genomics Proteomics. 2023;20(4):383–97. DOI: 10.21873/cgp.20390
8. Zhu G., Xiong Z., Chen W., Zhu Z., Wang W. Identification of key biomarkers and related immune cell infiltration in cervical cancer tissue based on bioinformatics analysis. Sci Rep. 2023;13(1):10121. DOI: 10.1038/s41598-023-37346-z
9. Beilerli A., Gareev I., Beylerli O., Yang G., Pavlov V., Aliev G., et al. Circular RNAs as biomarkers and therapeutic targets in cancer. Semin Cancer Biol. 2022;83:242–52. DOI: 10.1016/j.semcancer.2020.12.026
10. Sufianov A., Begliarzade S., Beilerli A., Liang Y., Ilyasova T., Beylerli O. Circular RNAs as biomarkers for lung cancer. Noncoding RNA Res. 2022;8(1):83–8. DOI: 10.1016/j.ncrna.2022.11.002
11. Beilerli A., Begliarzade S., Sufianov A., Ilyasova T., Liang Y., Beylerli O. Circulating ciRS-7 as a potential non-invasive biomarker for epithelial ovarian cancer: An investigative study. Noncoding RNA Res. 2022;7(3):197–204. DOI: 10.1016/j.ncrna.2022.07.004
12. Begliarzade S, Sufianov A, Ilyasova T, Shumadalova A, Sufianov R, Beylerli O, Yan Z. Circular RNA in cervical cancer: Fundamental mechanism and clinical potential. Noncoding RNA Res. 2023 Nov 18;9(1):116-124. DOI: 10.1016/j.ncrna.2023.11.009.
13. Zhang P., Chen M. Circular RNA Databases. Methods Mol Biol. 2021;2362:109–18. DOI: 10.1007/978-1-0716-1645-1_7
14. Panda A.C., Dudekula D.B., Abdelmohsen K., Gorospe M. Analysis of circular RNAs using the web tool circinteractome. Methods Mol Biol. 2018;1724:43–56. DOI: 10.1007/978-1-4939-7562-4_4
15. Luna Buitrago D., Lovering R.C., Caporali A. Insights into online microRNA bioinformatics tools. Noncoding RNA. 2023;9(2):18. DOI: 10.3390/ncrna9020018
16. Soares L.C., de Souza R.J., Oliveira M.A.P. Reviewing FIGO 2018 cervical cancer staging. Acta Obstet Gynecol Scand. 2023;102(12):1757–8. DOI: 10.1111/aogs.14667
17. Li K., Du Y., Li L., Wei D.Q. Bioinformatics approaches for anti-cancer drug discovery. Curr Drug Targets. 2020;21(1):3–17. DOI: 10.2174/138 9450120666190923162203
18. Huang M., He Y.R., Liang L.C., Huang Q., Zhu Z.Q. Circular RNA hsa_circ_0000745 may serve as a diagnostic marker for gastric cancer. World J Gastroenterol. 2017;23(34):6330–8. DOI: 10.3748/wjg.v23.i34.6330
19. Zhang C., Wang J., Wang H., Li J. Interference of the circular RNA sperm antigen with calponin homology and coiled-coil domains 1 suppresses growth and promotes apoptosis of breast cancer cells partially through targeting miR-1236-3p/Chromobox 8 pathway. Clin Breast Cancer. 2024;24(3):e138–51.e2. DOI: 10.1016/j.clbc.2023.11.009
20. Jiao J., Zhang T., Jiao X., Huang T., Zhao L., Ma D., et al. hsa_ circ_0000745 promotes cervical cancer by increasing cell proliferation, migration, and invasion. J Cell Physiol. 2020;235(2):1287–95. DOI: 10.1002/jcp.29045
21. Chen Y., Ling C., Xu Y., Liu J., Tang W. Evaluation of diagnostic and prognostic value of hsa_circ_0084927 and analysis of associated ceRNA network in colorectal cancer. Int J Gen Med. 2022;15:4357–77. DOI: 10.2147/IJGM.S355043
22. Shi P., Zhang X., Lou C., Xue Y., Guo R., Chen S. Hsa_ circ_0084927 regulates cervical cancer advancement via regulation of the miR-634/TPD52 Axis. Cancer Manag Res. 2020;12:9435–48. DOI: 10.2147/CMAR.S272478
23. Qiu F., Ou D., Tan H., Gao Y., Zi D. The circCDK17/miR-122-5p/ASF1B axis regulates the progression of cervical cancer. Histol Histopathol. 2023;38(3):359–71. DOI: 10.14670/HH-18-527
24. Bai H., Song M., Jiao R., Li W., Zhao J., Xiao M., et al. DUSP7 inhibits cervical cancer progression by inactivating the RAS pathway. J Cell Mol Med. 2021;25(19):9306–18. DOI: 10.1111/jcmm.16865
25. Bhattacharjee R., Das S.S., Biswal S.S., Nath A., Das D., Basu A., et al. Mechanistic role of HPV-associated early proteins in cervical cancer: Molecular pathways and targeted therapeutic strategies. Crit Rev Oncol Hematol. 2022;174:103675. DOI: 10.1016/j.critrevonc.2022.103675
26. Yang D., Fan L., Song Z., Fang S., Huang M., Chen P. The KMT1A/ TIMP3/PI3K/AKT circuit regulates tumor growth in cervical cancer. Reprod Biol. 2022;22(3):100644. DOI: 10.1016/j.repbio.2022.100644
27. Lee J.W., Lee J., Moon E.Y. HeLa human cervical cancer cell migration is inhibited by treatment with dibutyryl-cAMP. Anticancer Res. 2014;34(7):3447–55.
28. Mokoala K.M.G., Lawal I.O., Maserumule L.C., Bida M., Maes A., Ndlovu H., et al. Correlation between [68Ga]Ga-FAPI-46 PET Imaging and HIF-1α Immunohistochemical Analysis in Cervical Cancer: Proof-of-Concept. Cancers (Basel). 2023;15(15):3953. DOI: 10.3390/cancers15153953
29. Fan Y., Wang Y., Liu F., Wang H., Li Q. SEC61G promotes cervical cancer proliferation by activating MAPK signaling pathway. Dis Markers. 2022;2022:7016079. DOI: 10.1155/2022/7016079 30 Chandrasekar S.A., Palaniyandi T., Parthasarathy U., Surendran H., Viswanathan S., Wahab M.R.A., et al. Implications of Toll-like receptors (TLRs) and their signaling mechanisms in human cancers. Pathol Res Pract. 2023;248:154673. DOI: 10.1016/j.prp.2023.154673
30. Kang M., Qiu J., Wei H., Li J. A bibliometric analysis of global research trends of inflammation in cervical cancer: A review. Medicine (Baltimore). 2023;102(49):e36598. DOI: 10.1097/MD.0000000000036598
31. Zheng Y., Liu J., Beeraka N.M., Manogaran P., Vikram P.R.H., Yn L.D., et al. Inflammation and stem cell stochasticity of HPV-induced cervical cancer: epigenetics based biomarkers through microbiome and metabolome for personalized medicine: a systematic review. Curr Med Chem. 2023 Nov 24. DOI: 10.2174/0109298673257429231108 072717
Беглярзаде Сема Арзуман кызы — аспирант, кафедра онкологии, радиологии и радиотерапии
Тюмень
Тамразов Расим Ильхам оглы — д.м.н., профессор, кафедра онкологии, радиотерапии с курсом онкоурологии
Москва
Мусаев Эльмар Расим оглы — д.м.н., профессор, член-корр. РАН, кафедра онкологии
Москва
Вонг Чунлеи — профессор, отделение нейрохирургии
Харбин
Беглярзаде С.А., Тамразов Р.И., Мусаев Э.Р., Вонг Ч. Профиль экспрессии циркулярных РНК при раке шейки матки и построение регуляторной сети «циркулярные РНК — микроРНК — матричные РНК». Креативная хирургия и онкология. 2024;14(2):116-126. https://doi.org/10.24060/2076-3093-2024-14-2-116-126
Begliarzade S.A., Tamrazov R.I., Musaev E.R., Wang C. Circular RNA Expression Profile in Cervical Cancer and Construction of the Circular RNA‑MicroRNA‑Messenger RNA Regulatory Network. Creative surgery and oncology. 2024;14(2):116-126. (In Russ.) https://doi.org/10.24060/2076-3093-2024-14-2-116-126
Федеральное государственное бюджетное образовательное учреждение высшего образования «Башкирский государственный медицинский университет» Министерства здравоохранения Российской Федерации
450008, Республика Башкортостан, г. Уфа, ул. Пушкина, д. 96, корп. 98
Тел./факс: +7 (347) 273-56 -97
E-mail: csurgonco@bashgmu.ru






























